
Kinetics of non-catalytic synthesis of bis(2-ethylhexyl)sebacate at high pressures
Ram C.
Narayan
and
Giridhar
Madras
 *
*
Department of Chemical Engineering, Indian Institute of Science, Bangalore 560012, India. E-mail: giridhar@chemeng.iisc.ernet.in; Tel: +91 80 22932321
First published on 8th November 2016
Abstract
The bis(2-ethylhexyl)ester of sebacic acid is a widely used synthetic lubricant having various applications in the aerospace, automobile and manufacturing industries. This ester was synthesized directly from sebacic acid under subcritical and near critical conditions of 2-ethylhexanol without the addition of external catalysts for the first time. Reversible reaction kinetics was used to fit the experimental data considering the effect of acid catalysis across different initial molar ratios and temperatures. Equilibrium conversion was achieved within an hour of reaction even under subcritical conditions and at a lower temperature of 523 K. The synthesis route undertaken is proposed to have an economic as well as an environmental edge, due to lower molar excess of alcohol and shorter reaction time even without the addition of external catalysts.
1. Introduction
Breakdown products released due to increased loss of mineral oil based lubricants to the environment lead to aquatic toxicity and release of harmful volatile compounds into the atmosphere.1,2 The production of these conventional mineral oil based lubricants is not sustainable as it is dependent on fast depleting crude oil sources. The properties of the base fluid are not consistent and highly dependent on crude oil sources.3 Thus, there is an impetus towards developing synthetic and environmentally friendly lubricants that are produced from renewable agricultural sources. Esters can be tailored to cater to the ever increasing lubricating challenges and applications without compromising biodegradability and performance. The methodology may involve simple esterification4 or more complicated routes.5,6 Although vegetable oils (triglyceride esters) have been historically used as lubricants (especially castor oil and rapeseed oil) and have excellent biodegradability (90–98% (ref. 7)) and viscosity indices, they have shortcomings such as oxidative and hydrolytic stability.8Dibasic esters are an important class of these biolubricant esters.9 Conventional synthesis of diesters is based on enzyme10–13 or chemical catalysis.14–18 Many of these diesters are environmentally safe and have been tested for biodegradability.19 Among these, higher alcohol esters of dicarboxylic acids are specifically relevant as biolubricants. These esters have been synthesized using fatty acids, methyl esters or triglycerides as precursors by using catalytic means.20–25 Enzyme catalyzed processes are a greener alternative to chemical catalysts and are also highly selective. However, they are expensive, time consuming and sensitive to feedstock impurities. Although reports on catalyzed esterification are abundant in the literature, catalysts are not absolutely essential for esterification to occur. Esterification/transesterification can be effected by manipulating the phase of the reaction mixture alone. This is accomplished by increasing both the temperature and the pressure of the reactant system near to or above the critical point (supercritical state) of the alcohol (or the mixture), that changes the properties of the alcohol like the dielectric constant (and thus polarity), solubility parameter and density.26 For process operations, these fluids have advantages such as gas-like diffusivities and viscosities that enhance mass transfer but retain liquid like densities that help with solvation. More importantly, these properties can be tuned with temperature and pressure as parameters.
Conventional catalytic procedures are associated with acidic wastewater, neutralized salts, soaps or deactivated catalysts. These cause disposal problems and also reduce product purity. In reactions involving sub/supercritical alcohols, the necessity of adding a catalyst or a solvent may be obviated. The consequent simplicity of the downstream operations makes the process more economic and environmentally friendly (due to reduction in process wastes). Despite many advantages, certain shortcomings exist, such as harshness of operating conditions and requirement of high molar excess of reactants that affect process economics and safety.27,28 Most of these studies use supercritical alcohols and are focused on the production of fatty acid methyl or ethyl esters used as biodiesel synthesized from a wide variety of fatty acids and triglycerides.29–31 Comparatively, there are a few studies involving supercritical higher alcohols, like butanol,32 isoamyl alcohol33 and octanol.34 Non-catalytic reactions in supercritical fluids other than alcohols are still rare, like a recent study using diethyl ether.35 However, there are no reports on non-catalytic esterification of dicarboxylic acids with alcohols, specifically higher alcohols (under subcritical or supercritical conditions).
In this study, 2-ethylhexyl esters of sebacic acid were synthesized under subcritical and supercritical conditions of 2-ethylhexanol for the first time. These esters are applicable in the automobile and aerospace industries.14,36,37 Esterification was conducted at different temperatures and molar ratios up to a reaction time of 90 min. An analysis of possible phase conditions of the reaction mixture was made using suitable mixing rules. Further, the experimental conversion data were fitted to a second order reversible rate expression with rate constants determined under different reaction conditions.
2. Materials, methods and analysis
2.1. Materials
Dodecanedioic acid (sebacic acid) (>98% by GC) was purchased from SRL Chemicals. Methanol (>99.5% by GC), 2-ethylhexanol (>99% GC) and n-heptane (analytical grade) were purchased from Merck India Pvt Ltd. Butyl dodecanoate (>99% by GC) was obtained from Sigma Aldrich Ltd. (USA). All chemicals were used as received without further purification for all the reactions and subsequent chromatographic analyses.2.2. Methods
The esterification reactions were conducted in 10 cm3 stainless steel reactor tubes. One end of the tube is permanently sealed. The other end consists of a plug-nut–ferrule compression fitting for loading the reactants and collecting products after the reaction. The reactor tubes can withhold pressures up to 400 bar. SS-316 was used in fabricating all the parts of the reactor. The reactant mixture (sebacic acid and alcohol) was isochorically heated to the reaction temperature and pressure at which investigation is to be made. The reactant loading was calculated using a PR-EOS (with LB mixing rules38) based on the molar ratio at the start of the reaction. These calculations require pure component critical properties. The pure component critical properties for 2-ethylhexanol were taken from the NIST fluid property database. The critical properties for sebacic acid were estimated using suitable group contribution methods, as experimental determination of these properties is not possible due to chemical decomposition under critical conditions.39 Calculated quantities of sebacic acid and 2-ethylhexanol were loaded into the reactor tubes and placed in a furnace set at the desired temperature. The reaction time in all experiments was the time at which the reactors that were placed in the furnace attained the particular operating temperature. In order to stop further esterification, the reactor tubes were quenched in ice, which drastically reduced the temperature and pressure to ambient conditions. The conversions were estimated using GC (gas chromatography) analysis discussed in the succeeding section. The samples for these were obtained by suitable dilutions of the reaction mixture in n-heptane and by spiking a known quantity of butyl dodecanoate that acts as an internal standard (IS). The GC sample vials were centrifuged at 2000 rpm for 5 min to obtain a clear supernatant free from solids.2.3. Analysis
The reaction samples were analyzed by using gas chromatography (Varian CP 3800). The liquid sample was introduced into the injector port maintained at 300 °C. The components were separated on a capillary column (VF-5ms, 30 m × 0.25 mm with 0.25 μm film thickness of the 5% phenyl-methylpolysiloxane column) with the column oven maintained at 300 °C (isothermal program). Helium (99.9995%) was used as a carrier gas. The components were detected using a flame ionization detector (FID) maintained at 300 °C and fueled by pure hydrogen and oxygen gases (with pure nitrogen as the makeup gas). A linear calibration curve was obtained by injecting chemically synthesized purified esters. The global experimental error in the data corresponds to about ±3% of the yield based on triplicate experiments.3. Results and discussion
The critical temperature and pressure of pure 2-ethylhexanol are 641 K and 28 bar, respectively. The critical temperature of the alcohol is higher than methanol, ethanol or 1-butanol. On the contrary, the critical pressures are far lower than for the lower alcohols, obviating the need for very high operating pressures (200–400 bar) that are typical of non-catalytic biodiesel (fatty acid methyl/ethyl ester) synthesis. However, all the esterification reactions were conducted at a pressure of 60 bar, that is roughly twice the critical pressure of the alcohol. Beyond these pressures at constant temperature, the effect of pressure on conversion is minimal, as there is no significant change in density with pressure.40,41 The variation of conversion with time was studied at different molar ratios and temperatures. It should be noted that the reactor loading was calculated as discussed in section 2.2 and varies with molar ratio and temperature. The corresponding global densities (ratio of total weight of reactants and reactor volume) are given in Tables 1 and 2. Further, the experimental conversion data were fitted using reversible reaction kinetics that incorporates the contribution of carboxylic acid, i.e. sebacic acid towards the catalytic activity.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1
:1
| Temperature (T/K) | (k1 × 102)/L2 mol−2 s−1 | (k2 × 102)/L2 mol−2 s−1 | Density (ρ/kg m−3) |
|---|---|---|---|
| 523 | 0.41 | 1.51 | 676 |
| 573 | 0.68 | 2.36 | 619 |
| 623 | 1.04 | 3.55 | 542 |
| 673 | 2.05 | 7.49 | 441 |
| Activation energy (Ea/kJ mol−1) | 25 | 23 |
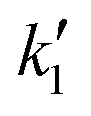 and
and 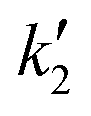 using eqn (5) and kf using eqn (6). Operating conditions: T = 623 K and P = 60 bar
using eqn (5) and kf using eqn (6). Operating conditions: T = 623 K and P = 60 bar
| Initial molar ratio | (k1 × 102)/L2 mol−2 s−1 | (k1 × 102)/L2 mol−2 s−1 | (kf × 102)/s−1 | (
|
(
|
Density(ρ/kg m−3) |
|---|---|---|---|---|---|---|
| 2:1 |
0.64 | 1.73 | — | 0.78 | 2.07 | 594 |
| 5:1 |
1.04 | 3.55 | — | 2.73 | 7.71 | 542 |
| 10:1 |
0.33 | 0 | 2.95 | — | — | 515 |
| 40:1 |
0.28 | 0 | 3 | — | — | 490 |
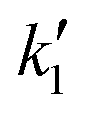
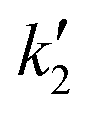
3.1. Effect of molar ratio
Molar ratio is one of the important variables affecting the equilibrium conversion of the esterification reaction. The effect of molar ratio was studied between 2:1 and 40:1, i.e. from stoichiometric amounts to systems with high molar excess of alcohol (diluted with sebacic acid). A constant temperature of 623 K and pressure of 60 bar were maintained. The corresponding variation of conversion with time is shown in Fig. 1(a). It was observed that different equilibrium conversions, 42% and 67%, are attained at molar ratios of 2:1 and 5:1, respectively. The lower equilibrium conversion is due to the enhancement of hydrolysis reaction at lower molar ratios. The variation of conversion with time for 10:1 and 40:1 molar ratios was very similar. At these high molar ratios, esterification is dominant over hydrolysis. This is because of the high molar excess of alcohol, which favors the forward reaction and hinders the backward reaction. Thus, the reaction rate (and thus conversion) is dependent solely on the concentration of sebacic acid and is nearly independent of the initial molar ratio (at higher molar ratios). The kinetic development using rate expressions to represent the experimental data is discussed in section 3.3. At stoichiometric amounts, although the equilibrium conversion was lower than those at other molar ratios, the reaction rate (at the start of reaction) was higher than those at molar ratios of 10:1 and 40:1, evident from the quick attainment of an equilibrium value of about 42% in 25 min. At a 5:1 molar ratio, an equilibrium conversion of about 68% was reached at about 40 min, with a superior reaction rate. However, at 10:1 and 40:1, high conversions up to 83% were attained within an hour, although the reaction rate is higher at lower molar ratios, especially during the initial reaction period.
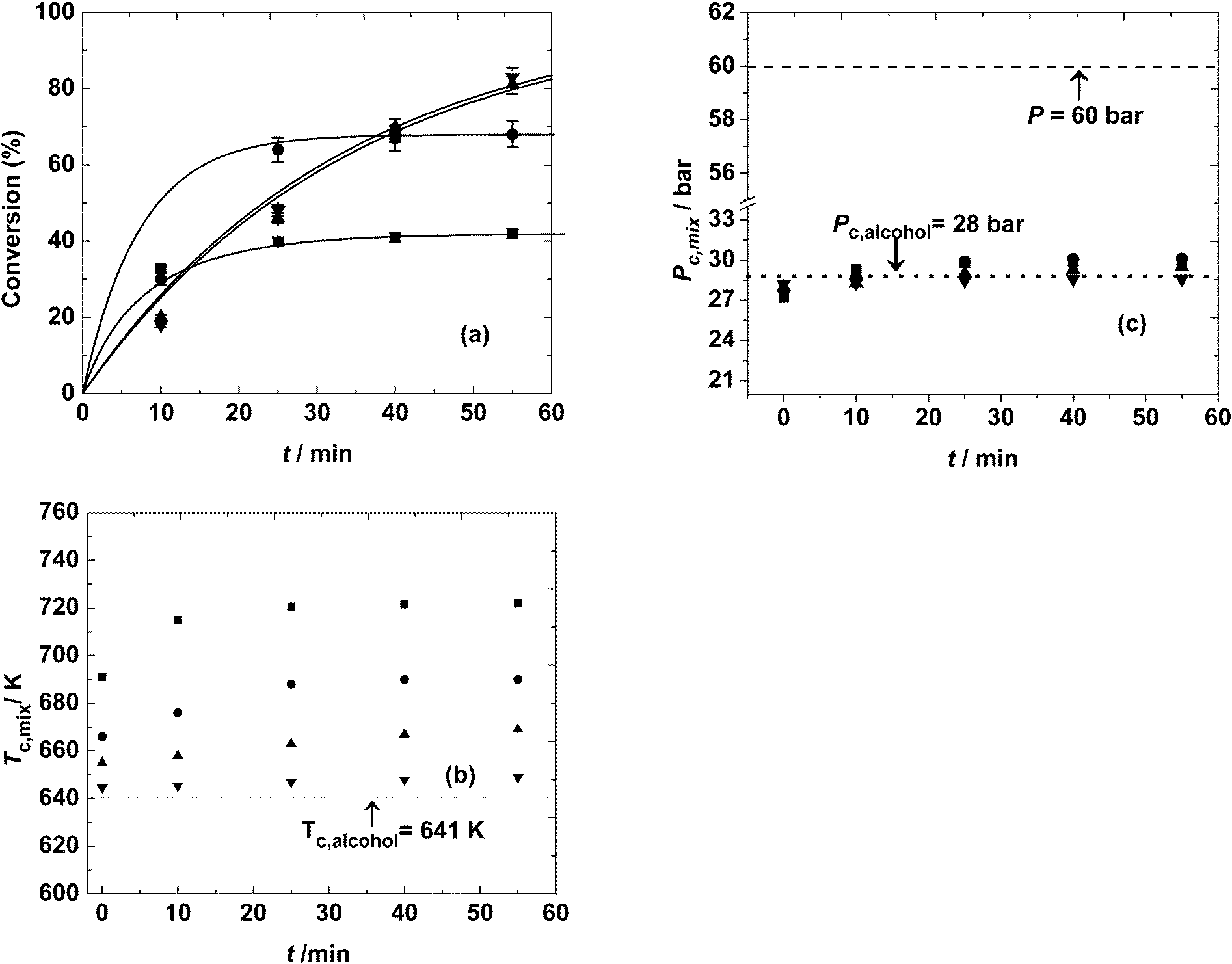 | ||
| Fig. 1 Variation of (a) conversion (%), (b) critical temperature of the mixture and (c) critical pressure of the mixture at molar ratios of 2:1 (■), 5:1 (●), 10:1 (▲) and 40:1 (▼) with reaction time. Operating conditions: T = 623 K and P = 60 bar. | ||
Sebacic acid is a solid with a high melting point of about 130 °C, while 2-ethylhexanol is practically a non-volatile non-polar liquid at room temperature. Under ambient conditions, these compounds are insoluble in each other. Thus, even catalytic reactions should be carried out at higher temperatures, preferably above the melting point of sebacic acid or should utilize suitable solvents to enhance phase miscibility or should use a different precursor such as dimethyl sebacate that is miscible with 2-ethylhexanol. Even under such conditions, the reaction time spans several hours.16 Another complication at higher temperature with the use of acid catalysts is the formation of ethers.42 The lower reaction rate can be attributed to the long carbon chain of 2-ethylhexanol and also branching which leads to increased steric hindrance.
The phase of the reaction mixture plays an important role in understanding reactions in supercritical fluids, as there is a marked change in the physicochemical properties around the critical point, for example, a decrease in dielectric constants and solubility parameters and liquid like densities at high temperatures (>473 K). In most cases, a high excess of alcohol is used to aid in the solvation of the triglyceride molecules as a triglyceride–methanol mixture is immiscible under ambient conditions. In such cases, the phase of the reaction mixture is not very different from the phase of the pure alcohol under the reaction conditions. When lower molar ratios are used, the phase of the reaction mixture is determined by both reactants, especially if the participating chemical species have very different critical points.
For the reaction system under consideration, the experimental data on the phase equilibria that would help discern the variation of critical points at different mixture compositions were unavailable. Experimental as well as modeling approaches for describing the phase equilibria of biodiesel–methanol–triglyceride systems are well investigated.43,44 However, only a few related studies of vapor–liquid equilibria exist which consider mixtures of small chain dicarboxylic acid esters, lower alcohols (methanol and ethanol) and water, useful in designing reactive distillation processes. Even these studies report equilibria data only up to the boiling point of the alcohol/water mixture.45–47 With the help of vapor–liquid equilibria, suitable interaction parameters can be calculated using the equation of state (EOS) approach to predict mixture critical points at different molar ratios. However, the critical point may be estimated directly using Lorentz–Berthelot (LB) mixing rules. These mixing rules are popularly used in reactions with supercritical alcohols.38,48 Using this approach, pseudo-critical temperature and pressure (of the reaction mixture) may be estimated. These mixing rules are well suited for components having unlike size and energy parameters and can be applied only to a pair of molecules at once.49,50 However, they provide good phase predictions and can be conveniently generalized for multicomponent mixtures.51 These mixing rules have also been successfully used in correlating phase compositions in mixtures of supercritical carbon dioxide and solid solutes.52,53
For estimating the critical point using LB-mixing rules, the pure component critical properties, viz. critical temperature, critical pressure, critical volume and critical compressibility factor, are required. Along with these, the composition of the reaction mixture is required. Using these mixing rules, the critical temperature and pressure of the mixture can be estimated for initial molar ratios between 2:1 and 40:1. The critical point of the mixture varies with the composition of the reaction mixture (that changes with reaction time). The variation of the critical temperature and pressure of the mixture with time for different molar ratios is shown in Fig. 1(b) and (c). It can be observed that the critical pressure of the mixture is almost invariant with molar ratio and is similar to the critical pressure of 2-ethylhexanol. This is because the critical pressure of sebacic acid (calculated by the Constantinou and Gani group contribution technique) is very similar to the critical pressure of 2-ethylhexanol. It is also because of the low critical molar volume of water. This variation is different from those observed in triglyceride–methanol or fatty acid–methanol mixtures, popularly used in synthesizing biodiesel, that have very different pure component critical pressures.54 However, the critical temperature of the mixture differs substantially with molar ratio, being the highest at the lowest molar ratio. It also increases with reaction time, due to the formation of a diester that has a high critical temperature. The difference between the critical temperatures of the mixture between the conditions at the onset of the reaction and the state of equilibrium is most marked at the lowest molar ratio of 2:1 (up to 80 K). At higher molar ratios of 10:1 and 40:1, the critical point was not very different from that of 2-ethylhexanol and the phase considerations do not vary with the progress of the reaction. Thus, at 623 K, where the variation of molar ratio was studied, the reaction mixture is expected to be in liquid phase throughout the reaction.
3.2. Effect of temperature
The effect of temperature was studied at a molar ratio of 5:1 (of 2-ethylhexanol:sebacic acid) at temperatures of 523–673 K and a constant pressure of 60 bar. This molar ratio was chosen due higher equilibrium conversion (as compared to 2:1) and reaction rate. The higher molar ratios of 10:1 and 40:1 were not considered due to a lower reaction rate, especially at the beginning of the reaction, although high conversions were ultimately attained at longer reaction times. Further, lower molar ratios have economic and environmental benefits.55
Fig. 2(a) shows the variation of critical temperature and pressure of mixtures at different temperatures with time. The system reached an equilibrium value of 68% that was lesser than complete conversion, due to the lower molar ratio used and prevalence of backward reaction. At 673 K, equilibrium conversion was reached within 15 min. Even at a lower temperature of 523 K, the equilibrium was reached within an hour of reaction time.
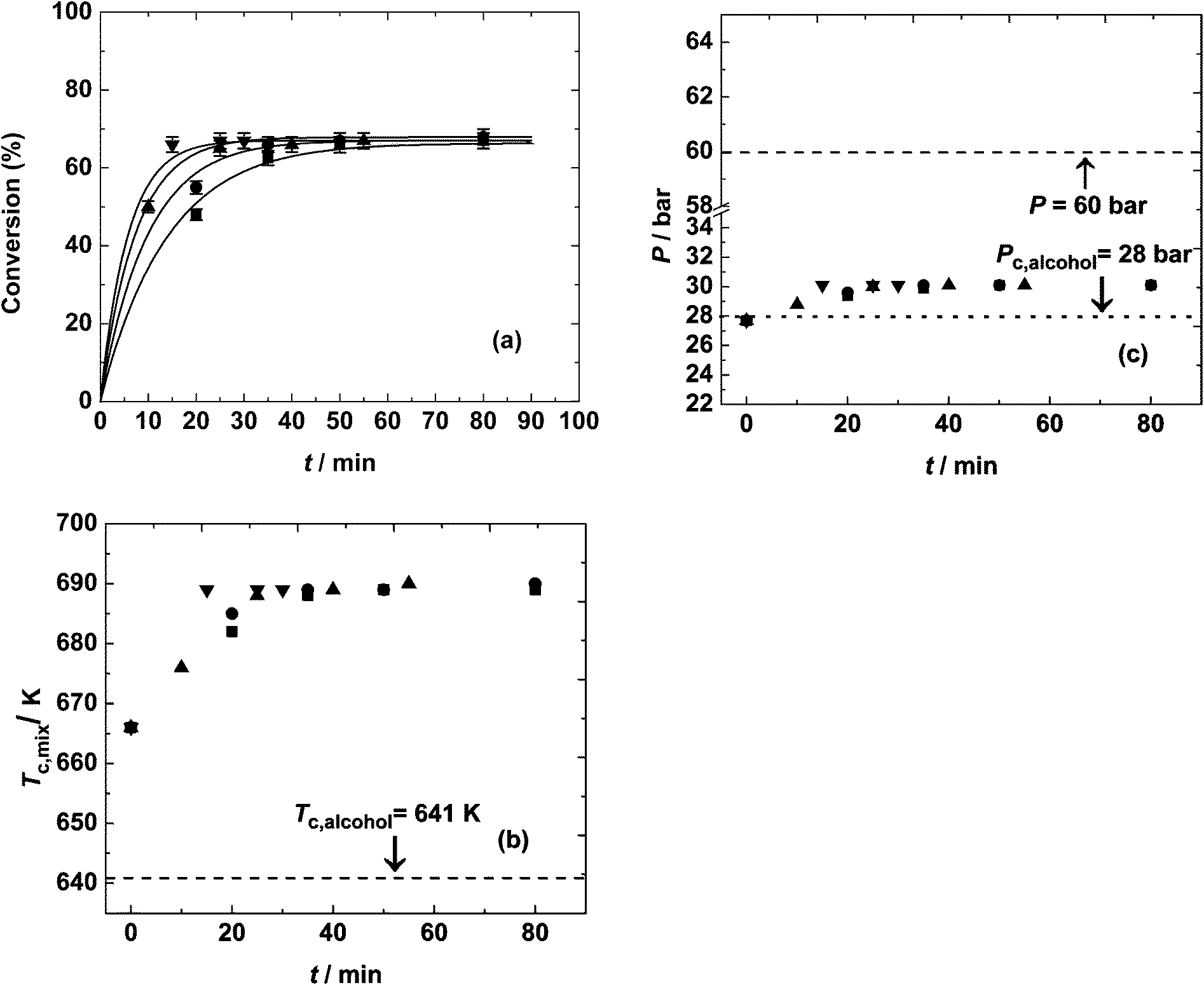 | ||
| Fig. 2 Variation of (a) conversion (%), (b) critical temperature of the mixture and (c) critical pressure of the mixture at different temperatures: ■, 523 K; ●, 573 K; ▲, 623 K and ▼, 673 K. Operating conditions: P = 60 bar and initial molar ratio of 5:1. | ||
There is a variation of the critical point of the mixture with reaction time even at a fixed initial molar ratio (in this case 5:1) because of the change in composition of the reaction mixture with time. This variation can be neglected for higher molar ratios (such as 40:1) as the critical point is close to that of 2-ethylhexanol (as shown in Fig. 1(b)), unlike at 2:1 and 5:1 initial molar ratios. The critical point variation with progress of the reaction is shown in Fig. 2(b) and (c). As conversion increases, the concentration of the diester, bis(2-ethylhexyl)sebacate, increases. This ester has a higher critical temperature than that of the precursor sebacic acid due to the high molecular weight, while the formed water has a critical temperature similar to that of 2-ethylhexanol that is being consumed. Thus, the critical temperature of the mixture increases with time until equilibrium is reached. However, the critical pressure of the mixture remains almost constant throughout the reaction, due to similar critical pressures of the reactants and also due to the smaller volumetric contribution of water. The critical temperature and pressure of the mixture at equilibrium are 689 K and 30 bar, respectively, at an initial molar ratio of 5:1.
In the temperature range investigated, the reactions took place in the subcritical liquid phase (as the operating pressure was more than the critical pressure of the mixture), except for a brief initial period at a temperature of 673 K, as it was just above the critical temperature at the onset of the reaction (of 666 K). A faster rate in this period was also observed up to an equilibrium conversion of 68%, after which the system is again under sub-critical conditions. Even at 673 K, although the reaction mixture was above the critical point of 2-ethylhexanol, it was in the subcritical state due to the low molar ratio (5:1) due to the increased contribution of sebacic acid and the quickly formed diester.
At 673 K, beyond 30 min reaction time, degradation was observed. Even at 623 K, degradation occurred after an hour of reaction, at all molar ratios between 2:1 and 40:1. Temperatures above 673 K were not investigated, due to the shift in the pyrolytic regime, that leads to the formation of a mixture of undesired compounds, not useful as lubricants. The variation of equilibrium conversion with temperature was not considerable in the temperature range investigated. This could mainly be due to the subcritical conditions of the reaction mixtures. In a recent work, SOOD (soybean oil deodorizer distillate) was converted to ethyl esters using supercritical ethanol, at a 7:1 initial molar ratio and 20 MPa. It was observed that the equilibrium conversion was about 85% and did not vary with temperatures between 523 K and 600 K.56 This was also observed in other esterification systems, both catalytic and non-catalytic (supercritical).31,57–59 This can be attributed to the low heat of reaction for esterification58,60 which would make the equilibrium conversion almost temperature invariant.
3.3. Reaction kinetics
Esterification/transesterification is modeled using reversible kinetics at lower molar ratios due to the prevalence of backward/hydrolysis reaction. An equilibrium conversion is attained after a particular reaction time, reaching nearly complete conversions only when high molar excess of alcohol is used or if water formed is continuously removed from the reaction mixture. Consider the following reaction scheme, where sebacic acid (A) reacts with 2-ethylhexanol (B) to give bis(2-ethylhexyl)sebacate (E) and water (W).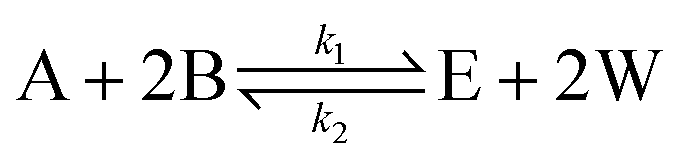 | (1) |
If the concentrations of these species are denoted by CA, CB, CE and CW and the rate constants for forward and backward reactions are denoted by k1 and k2, respectively, then the variation of the concentration of A with time is given by
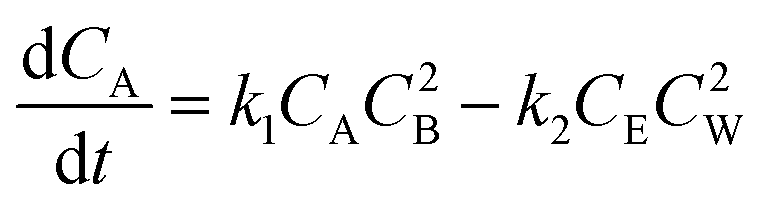 | (2) |
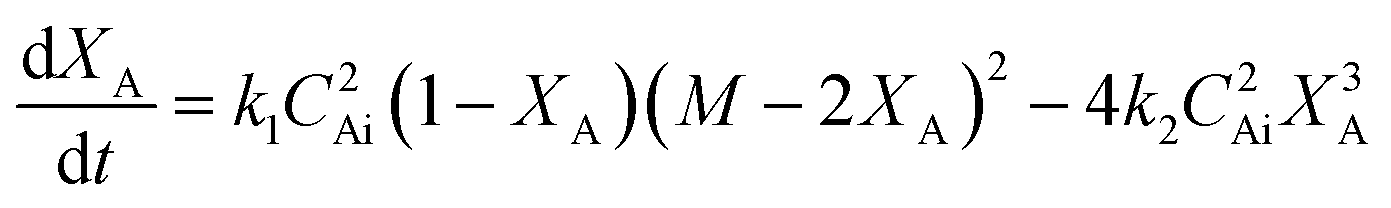 | (3) |
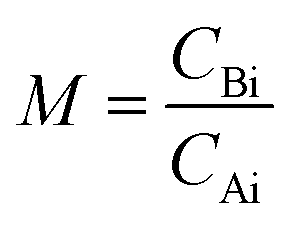 is the initial molar ratio of 2-ethylhexanol to sebacic acid and CAi and CBi are the initial concentrations of sebacic acid and 2-ethylhexanol, respectively.
is the initial molar ratio of 2-ethylhexanol to sebacic acid and CAi and CBi are the initial concentrations of sebacic acid and 2-ethylhexanol, respectively.
Although sebacic acid is not as acidic as mineral acids, at higher sebacic acid loading, the reaction could be catalyzed by sebacic acid itself. The variation of concentration of A would then be given by eqn (4). Similar kinetic expressions for supercritical esterification of fatty acids with methanol have been derived earlier to account for the contribution of catalysis by carboxylic acid.61
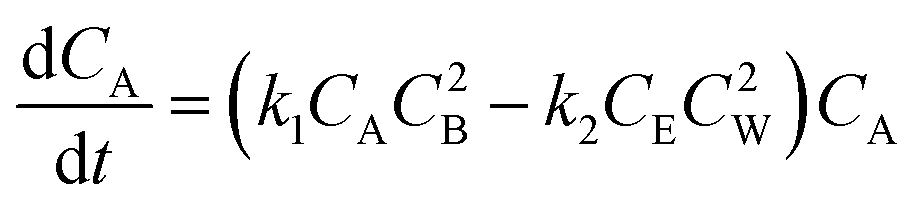 | (4) |
In terms of conversion of sebacic acid with time, eqn (4) can be rewritten as
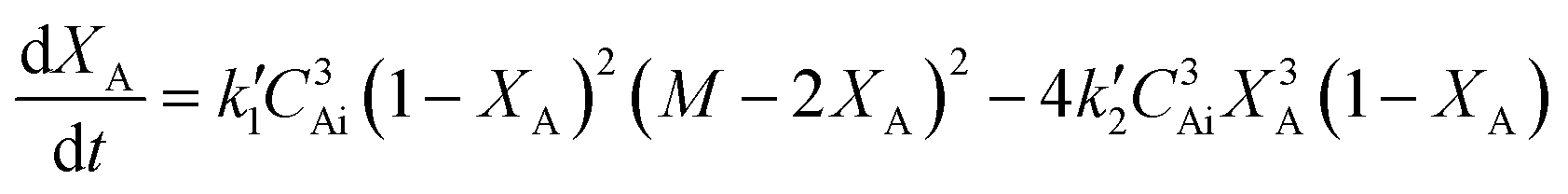 | (5) |
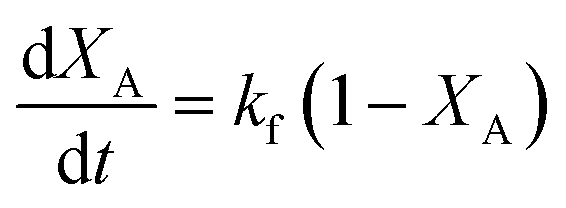 | (6) |
The experimental data for variation of conversion of sebacic acid at different initial molar ratios and temperatures with reaction time were fitted using the kinetic models described by eqn (3), (5) and (6). At 623 K, for molar ratios of 2:1 and 5:1, eqn (3) and (5) were used to estimate the rate constants (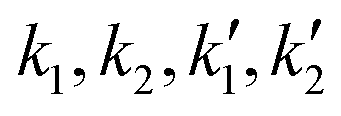 ). These are tabulated in Tables 1 and 2. In the case of a 5:1 molar ratio, eqn (3) gave a better fit (R2 = 0.993) to the experimental conversion data than eqn (5) (R2 = 0.94). Eqn (5), which included a term for sebacic acid catalytic contribution, overestimated the equilibrium conversion. In the case of a 2:1 molar ratio, eqn (3) underestimates the initial conversion values unlike eqn (5) that provides a closer fit to the experimental data. This is because of the higher initial rate as predicted by eqn (5) as compared to eqn (3). Thus, acid catalysis by sebacic acid is more prominent at 2:1 than at 5:1. At molar ratios of 10:1 and 40:1, the estimated value of the hydrolysis rate constants
). These are tabulated in Tables 1 and 2. In the case of a 5:1 molar ratio, eqn (3) gave a better fit (R2 = 0.993) to the experimental conversion data than eqn (5) (R2 = 0.94). Eqn (5), which included a term for sebacic acid catalytic contribution, overestimated the equilibrium conversion. In the case of a 2:1 molar ratio, eqn (3) underestimates the initial conversion values unlike eqn (5) that provides a closer fit to the experimental data. This is because of the higher initial rate as predicted by eqn (5) as compared to eqn (3). Thus, acid catalysis by sebacic acid is more prominent at 2:1 than at 5:1. At molar ratios of 10:1 and 40:1, the estimated value of the hydrolysis rate constants 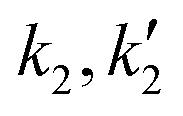 were zero, which is to be expected due to the high molar excess of alcohol in the reaction mixture, which practically makes it an irreversible reaction. At these molar ratios, eqn (5) reduced to a second order (with respect to fatty acid) like rate expression, which did not fit the conversion data because the acid catalytic activity is negligible due to dilute concentrations of sebacic acid. Eqn (5) gives second order fits that underestimate conversion values especially at longer reaction times.
were zero, which is to be expected due to the high molar excess of alcohol in the reaction mixture, which practically makes it an irreversible reaction. At these molar ratios, eqn (5) reduced to a second order (with respect to fatty acid) like rate expression, which did not fit the conversion data because the acid catalytic activity is negligible due to dilute concentrations of sebacic acid. Eqn (5) gives second order fits that underestimate conversion values especially at longer reaction times.
However, at higher molar ratios of 10:1 and 40:1, the assumption of a pseudo-first order reaction (eqn (6)) holds true. The corresponding pseudo first order rate constants (kf) (calculated from eqn (6)) are shown in Table 1. The pseudo first order rate constants were similar at both these initial molar ratios. Thus, in Fig. 1(a), for molar ratios of 5:1, 10:1 and 40:1, the conversion data were fitted using eqn (3), and eqn (5) was used to fit a 2:1 molar ratio. The lines in Fig. 2(a) showing the variation of conversion at a 5:1 molar ratio across different temperatures (523–673 K) were also fitted using eqn (3). The corresponding rate constants are shown in Table 2. Nearly a fivefold increase was observed in the value of both the rate constants (k1, k2), upon raising the temperature of the reaction system from 523 K to 673 K. As the initial molar ratio is just 2.5 times the stoichiometric amounts, the rate constant for hydrolysis was higher than that for esterification. The subcritical conditions of the reaction mixture (except in the brief initial period at 673 K) might be another reason for this.
Kinetic data for synthesis of diesters are not commonly reported in the literature, even though variation of experimental conversion data with time was plotted. Even among these, the kinetic parameters for synthesis of lower dicarboxylic acids (like adipic acid57 or succinic acid62) with lower alcohols were reported. The rate constants were fitted using the Arrhenius equation, to determine the activation energy of the reaction. At a 5:1 molar ratio and a temperature range of 523–623 K, activation energies of 25 kJ mol−1 and 23 kJ mol−1 were estimated for the esterification and hydrolysis reaction, respectively. The similarity in activation energies may be due to the subcritical conditions of the reaction mixture and low heat of esterification. The corresponding Arrhenius plot is shown in Fig. 3(a). There is a slight jump in the value of the rate constants at 673 K, as the reaction mixture was around/closer to the critical point of the reaction mixture. However, when the logarithms of these rate constants were plotted against global density (the ratio of total weight of reactants loaded to the volume of the reactor), as shown in Fig. 3(b), a single straight line was obtained. This phenomenon was also observed in the degradation of certain polymers in supercritical fluids.63,64 Thus, the temperature and, indirectly, the density of the reaction mixture affect the rate of reaction.
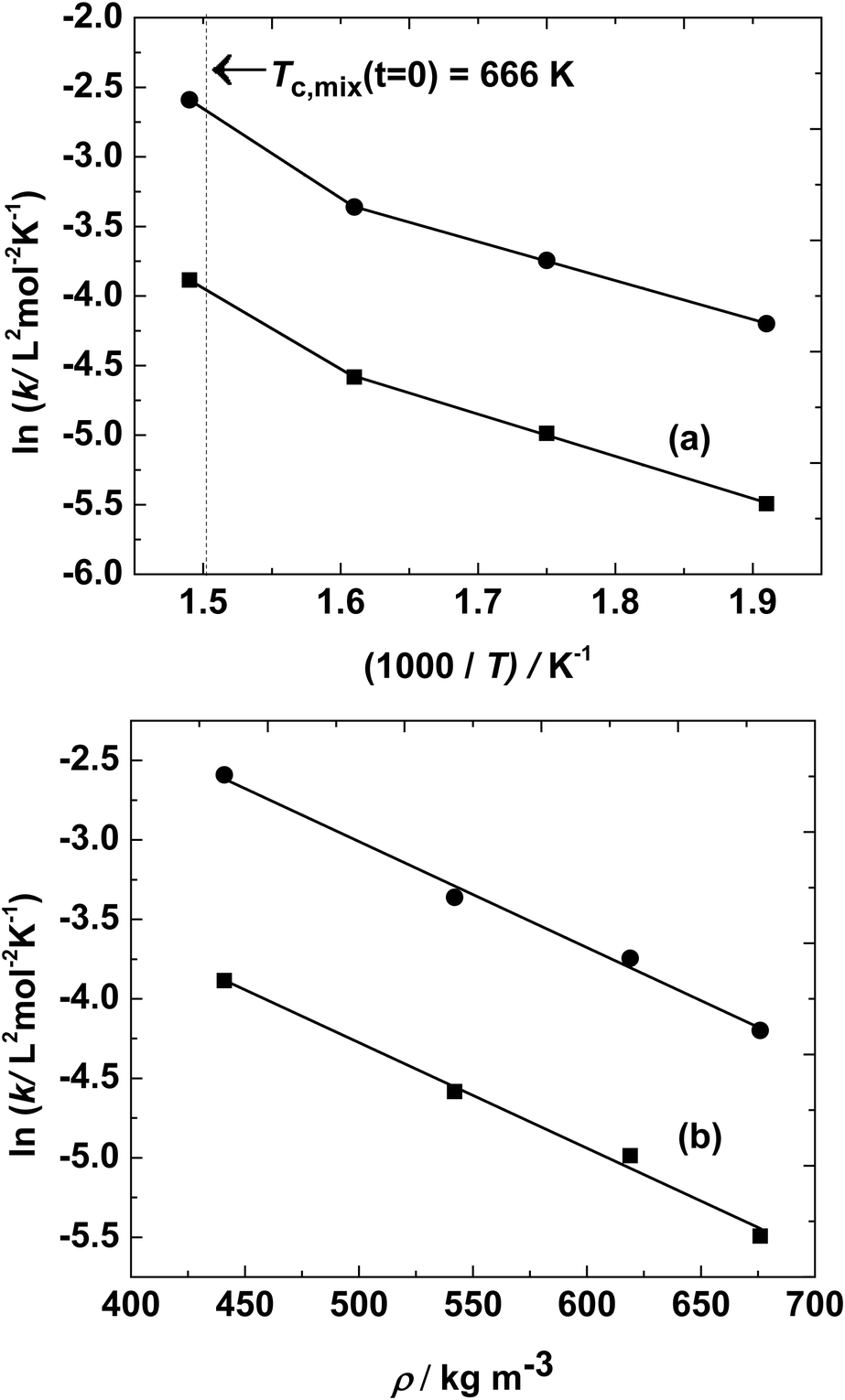 | ||
| Fig. 3 Variation of lnk1 (■) and lnk2 (●) with (a) temperature (T) and (b) density of reaction mixture (ρ). ‘k’ shown in the figure represents either k1 or k2. | ||
Reactions at temperatures above 673 K (especially greater than 693 K, which is the critical temperature of the mixture at 68% conversion and 5:1 initial molar ratio) were not investigated as pyrolytic regime predominates, leading to higher errors in estimating the rate constants. Possible reactions such as thermal cracking, decarboxylation and hydrolysis can be expected to occur in this regime, similar to those obtained with biodiesel synthesis.29 Further, these reaction products are not suitable to be used as lubricants as they are of lower molecular weight and possess different functionality. The activation energy was lower than those of the zinc soap catalyzed reactions of fatty acid with hexanol (66 kJ mol−1).65 The obtained activation energies were lesser than those reported for diesters synthesized using catalysts (34 kJ mol−1 and 66 kJ mol−1).57,62 The value of activation energy was found to be similar for both esterification and hydrolysis reaction. These reaction systems are different as the reactants are highly polar and soluble in each other, unlike sebacic acid and 2-ethylhexanol (insoluble under ambient conditions) that are investigated in this study. Conversions up to 94% were obtained using lipase catalyzed (Novozym-435) synthesis of bis(2-ethylhexyl)sebacate from dimethyl sebacate with a reaction time of 20 h.11 In this system, double the stoichiometric excess of alcohol was taken. The methanol formed was removed by azeotropic distillation with isooctane to drive the forward reaction.
Transesterification was attempted without the use of catalysts using dimethyl sebacate and 2-ethylhexanol at 623 K and 60 bar. The conversions were below 2% even after one hour of reaction time (higher reaction times greater than this were not investigated due to potential thermal degradation). This can be attributed to the low reactivity of 2-ethylhexanol, even though 2-ethylhexanol and dimethyl sebacate are miscible. Transesterification is very successful with supercritical alcohols with lower number of carbon atoms (like methanol and ethanol). However, 2-ethylhexanol due to its lengthy carbon chain forms a weak nucleophile (unlike methanol and ethanol that are stronger nucleophiles) in attacking molecules of dimethyl sebacate. In addition, the branching of the alcohol may further decrease the reactivity due to an increase in steric hindrance. Sebacic acid as a precursor could act as an acidic catalyst (furthermore due to the presence of two carboxylic groups). The reactivity of sebacic acid with 2-ethylhexanol could be due to the formation of certain reactive hydrophilic aggregates. These require intermolecular hydrogen bonding between sebacic acid and 2-ethylhexanol, that would not be possible with the use of dimethyl sebacate. This kind of structuration was hypothesized in an earlier study, in which 2-ethylhexanol was reacted with oleic acid at 473 K. High temperatures break the hydrogen bonds between the fatty acid molecules (in this case intramolecular bonding between sebacic acid molecules), and the alcohol moiety is inserted into these active aggregates to form esters. The formation of water during esterification and the surface active properties of the ester and 2-ethylhexanol were supposed to stabilize these structures, resulting in high reactivity.21
The proposed route is superior to other methods, in that it uses the acid directly as the precursor (instead of dimethyl sebacate), thereby avoiding an additional methyl esterification step. The use of sebacic acid makes the purification easier, as the unreacted acid precipitates as a solid after the reaction mixture is brought back to ambient conditions. There are no by-products and toxic wastes generated in this route. The pressure of the reaction mixture is also much lower as compared with other supercritical biodiesel synthesis procedures that are operated at 4–8 times the operating pressures used in this study. In addition, the separation of water formed after the reaction is straightforward by simple gravity based settling under ambient conditions. This avoids solubility issues/emulsification (leading to a decrease in product purity) that could occur with an alkaline catalyst.16
The mixture of 2-ethylhexanol and bis(2-ethylhexyl)sebacate can be possibly purified using supercritical carbon dioxide based processes, rather than using conventional column chromatography procedures that are based on using non-renewable solvents like n-hexane and diethyl ether.14 This is due to the differences in pure component solubilities of at least an order of magnitude between bis(2-ethylhexyl)sebacate and 2-ethylhexanol.66,67 Alternatively, the reaction mixture could be purified by using supercritical carbon dioxide by continuously removing water to enhance the equilibrium conversion of the diester.68
However, the designing of the extraction/purification operation requires mixture solubilities that are not available in the literature. The molecular structures of the reactive hydrophilic aggregates could be characterized by using an in situ FTIR or a view cell that could withstand higher pressures. These could be the scope for a future study.
4. Conclusions
Bis(2-ethylhexyl)sebacate, a synthetic biolubricant diester, was synthesized from sebacic acid and 2-ethylhexanol in a single step without using catalysts. The experimental conversion data were fitted using second order reversible kinetics by also considering the effect of acid catalysis at lower molar ratios. Conversion data were similar at 10:1 and 40:1 molar ratios and can be modeled using a pseudo first order assumption. The highest conversion of 83% was obtained at 623 K and a 10:1 molar ratio in an hour. However, equilibrium conversion of 68% was attained even at the lowest investigated temperature of 523 K and an initial molar ratio of 5:1, within an hour and with a high reaction rate. The effect of temperature on equilibrium conversion was minimal at a 5:1 initial molar ratio possibly due to lower heat of esterification. The effect of initial molar ratio was however more profound in affecting the equilibrium conversion. The activation energies estimated using the Arrhenius equation were similar for both esterification and hydrolysis reaction (around 25 kJ mol−1). The use of the methyl ester of sebacic acid (instead of sebacic acid itself) as a precursor for transesterification was not successful and leads to abysmally low conversions. The reaction mixture was mostly in the subcritical liquid phase for all the temperatures and molar ratios investigated, except for a brief initial period of reaction at 673 K. The critical pressure of the mixture is not very different from the critical pressure of alcohol, with the progress of the reaction under different operating conditions. However, the critical temperature of the mixture varied substantially with the progress of reaction at different operating conditions.
Acknowledgements
The authors thank the Council of Scientific and Industrial Research (CSIR/395) for their financial support. The corresponding author thanks the Department of Science and Technology (DST), India for the J. C. Bose fellowship.References
- J. A. C. da Silva, A. C. Habert and D. M. G. Freire, Lubr. Sci., 2013, 25, 53–61 Search PubMed.
- H. Wagner, R. Luther and T. Mang, Appl. Catal., A, 2001, 221, 429–442 Search PubMed.
- P. Nagendramma and S. Kaul, Renewable Sustainable Energy Rev., 2012, 16, 764–774 Search PubMed.
- A. Chowdhury, R. Chakraborty, D. Mitra and D. Biswas, Ind. Crops Prod., 2014, 52, 783–789 Search PubMed.
- T. Mang and W. Dresel, Lubricants and lubrication, John Wiley & Sons, Weinheim, 2007 Search PubMed.
- S. Li, L. Bouzidi and S. S. Narine, Ind. Eng. Chem. Res., 2014, 53, 12339–12354 Search PubMed.
- H. M. Mobarak, E. Niza Mohamad, H. H. Masjuki, M. A. Kalam, K. A. H. Al Mahmud, M. Habibullah and A. M. Ashraful, Renewable Sustainable Energy Rev., 2014, 33, 34–43 CrossRef CAS.
- S. Z. Erhan, B. K. Sharma, Z. Liu and A. Adhvaryu, J. Agric. Food Chem., 2008, 56, 8919–8925 Search PubMed.
- J. Salimon, N. Salih and B. M. Abdullah, Int. J. Chem. Eng., 2012, 2012, 10 Search PubMed.
- S. Gryglewicz, J. Mol. Catal. B: Enzym., 2001, 15, 9–13 Search PubMed.
- S. Gryglewicz, Enzyme Microb. Technol., 2003, 33, 952–957 Search PubMed.
- M. B. Abdul Rahman, N. Chaibakhsh, M. Basri, A. B. Salleh and R. N. Z. R. Abdul Rahman, Appl. Biochem. Biotechnol., 2009, 158, 722–735 Search PubMed.
- A. E. Petersson, L. M. Gustafsson, M. Nordblad, P. Börjesson, B. Mattiasson and P. Adlercreutz, Green Chem., 2005, 7, 837–843 Search PubMed.
- S. Sunitha, S. Kanjilal, P. S. Reddy and R. B. N. Prasad, Tetrahedron Lett., 2007, 48, 6962–6965 Search PubMed.
- S. Gryglewicz, Synth. Lubr., 2000, 17, 191–200 Search PubMed.
- S. Gryglewicz, Appl. Catal., A, 2000, 192, 23–28 Search PubMed.
- S. Y. Chin, M. A. A. Ahmad, M. R. Kamaruzaman and C. K. Cheng, Chem. Eng. Sci., 2015, 129, 116–125 Search PubMed.
- Y.-T. Tsai, H.-M. Lin and M.-J. Lee, J. Taiwan Inst. Chem. Eng., 2011, 42, 271–277 Search PubMed.
- S. Gryglewicz and B. Kolwzan, Synth. Lubr., 2004, 20, 281–288 Search PubMed.
- L. Jassim, R. Yunus, U. Rashid, S. A. Rashid, M. A. Salleh, S. Irawan and F. Ghaemi, Chem. Eng. Commun., 2016, 203, 463–470 Search PubMed.
- C. Lacaze-Dufaure and Z. Mouloungui, Appl. Catal., A, 2000, 204, 223–227 Search PubMed.
- M. Lämsä, A. Huhtala, Y.-Y. Linko and P. Linko, Biotechnol. Tech., 1994, 8, 451–456 Search PubMed.
- E. Kleinaitė, V. Jaška, B. Tvaska and I. Matijošytė, J. Cleaner Prod., 2014, 75, 40–44 CrossRef.
- R. M. A. Saboya, J. A. Cecilia, C. García-Sancho, F. M. T. Luna, E. Rodríguez-Castellón and C. L. Cavalcante Jr, Appl. Clay Sci., 2016, 124–125, 69–78 Search PubMed.
- C. Xie, H. Li, L. Li, S. Yu and F. Liu, J. Hazard. Mater., 2008, 151, 847–850 Search PubMed.
- G. Madras, C. Kolluru and R. Kumar, Fuel, 2004, 83, 2029–2033 Search PubMed.
- A. Demirbas, Energy Explor. Exploit., 2007, 25, 63–70 CrossRef CAS.
- A. Demirbas, Energy Convers. Manage., 2008, 49, 125–130 CrossRef CAS.
- R. Sawangkeaw, S. Teeravitud, K. Bunyakiat and S. Ngamprasertsith, Bioresour. Technol., 2011, 102, 10704–10710 CrossRef CAS PubMed.
- C.-Y. Wei, T.-C. Huang and H.-H. Chen, J. Chem., 2013, 789594 Search PubMed.
- D. Yujaroen, M. Goto, M. Sasaki and A. Shotipruk, Fuel, 2009, 88, 2011–2016 Search PubMed.
- Y. Sun, S. Ponnusamy, T. Muppaneni, H. K. Reddy, P. D. Patil, C. Li, L. Jiang and S. Deng, Fuel, 2014, 135, 522–529 Search PubMed.
- R. C. Narayan and G. Madras, Energy Fuels, 2016, 30, 4104–4111 Search PubMed.
- Y. Warabi, D. Kusdiana and S. Saka, Bioresour. Technol., 2004, 91, 283–287 Search PubMed.
- H. Ulusal, G. Fındıkkıran, O. Demirkol, D. Akbaşlar and E. S. Giray, J. Supercrit. Fluids, 2015, 105, 146–150 Search PubMed.
- S. Gryglewicz, A. Stankiewicz, F. A. Oko and I. Surawska, Tribol. Int., 2006, 39, 560–564 Search PubMed.
- S. Gryglewicz and F. A. Oko, Ind. Lubr. Tribol., 2005, 57, 128–132 Search PubMed.
- K. Bunyakiat, S. Makmee, R. Sawangkeaw and S. Ngamprasertsith, Energy Fuels, 2006, 20, 812–817 Search PubMed.
- L. Constantinou and R. Gani, AIChE J., 1994, 40, 1697–1710 Search PubMed.
- J.-J. Jiang and C.-S. Tan, J. Taiwan Inst. Chem. Eng., 2012, 43, 102–107 Search PubMed.
- K. White, N. Lorenz, T. Potts, W. R. Penney, R. Babcock, A. Hardison, E. A. Canuel and J. A. Hestekin, Fuel, 2011, 90, 3193–3199 Search PubMed.
- R. K. Raut, M. Shaikh and S. Darbha, J. Chem. Sci., 2014, 126, 997–1003 Search PubMed.
- P. Hegel, G. Mabe, S. Pereda and E. A. Brignole, Ind. Eng. Chem. Res., 2007, 46, 6360–6365 Search PubMed.
- A. Velez, S. Pereda and E. A. Brignole, J. Supercrit. Fluids, 2010, 55, 643–647 Search PubMed.
- S.-B. Hung, I. K. Lai, H.-P. Huang, M.-J. Lee and C.-C. Yu, Ind. Eng. Chem. Res., 2008, 47, 3076–3087 Search PubMed.
- S.-B. Hung, H.-M. Lin, C.-C. Yu, H.-P. Huang and M.-J. Lee, Fluid Phase Equilib., 2006, 248, 174–180 Search PubMed.
- E. İnce and Ş. İ. Kırbaşlar, J. Chem. Thermodyn., 2003, 35, 1671–1679 Search PubMed.
- I. M. Lokman, M. Goto, U. Rashid and Y. H. Taufiq-Yap, Chem. Eng. J., 2016, 284, 872–878 CrossRef CAS.
- C. McCabe, A. Gil-Villegas and G. Jackson, J. Phys. Chem. B, 1998, 102, 4183–4188 Search PubMed.
- S. M. Walas, Phase equilibria in chemical engineering, Butterworth-Heinemann, Boston, 2013 Search PubMed.
- Y. Iwai, H. Uchida, Y. Koga, Y. Arai and Y. Mori, Ind. Eng. Chem. Res., 1996, 35, 3782–3787 Search PubMed.
- M. Gordillo, M. Blanco, A. Molero and E. Martinez De La Ossa, J. Supercrit. Fluids, 1999, 15, 183–190 Search PubMed.
- M. D. Gordillo, C. Pereyra and E. J. Martínez de la Ossa, J. Supercrit. Fluids, 2003, 27, 31–37 CrossRef CAS.
- P. Olivares-Carrillo, J. Quesada-Medina, A. Pérez de los Ríos and F. J. Hernández-Fernández, Chem. Eng. J., 2014, 241, 418–432 Search PubMed.
- C. Kiwjaroun, C. Tubtimdee and P. Piumsomboon, J. Cleaner Prod., 2009, 17, 143–153 Search PubMed.
- L. J. Visioli, F. de Castilhos, L. Cardozo-Filho, B. T. F. de Mello and C. da Silva, Fuel Process. Technol., 2016, 149, 326–331 Search PubMed.
- K.-W. Chan, Y.-T. Tsai, H.-M. Lin and M.-J. Lee, J. Taiwan Inst. Chem. Eng., 2010, 41, 414–420 Search PubMed.
- A. Alegría and J. Cuellar, Appl. Catal., B, 2015, 179, 530–541 Search PubMed.
- A. W. Go, P. L. Tran Nguyen, L. H. Huynh, Y.-T. Liu, S. Sutanto and Y.-H. Ju, Energy, 2014, 70, 393–400 Search PubMed.
- A. Chowdhury, D. Sarkar and D. Mitra, Chem. Eng. Technol., 2016, 39, 730–740 Search PubMed.
- E. Minami and S. Saka, Fuel, 2006, 85, 2479–2483 Search PubMed.
- A. K. Kolah, N. S. Asthana, D. T. Vu, C. T. Lira and D. J. Miller, Ind. Eng. Chem. Res., 2008, 47, 5313–5317 Search PubMed.
- G. Sivalingam and G. Madras, Ind. Eng. Chem. Res., 2002, 41, 5337–5340 Search PubMed.
- A. Marimuthu and G. Madras, Ind. Eng. Chem. Res., 2007, 46, 15–21 Search PubMed.
- E. J. M. de Paiva, M. L. Corazza, M. R. Sierakowski, J. Wärnå, D. Y. Murzin, F. Wypych and T. Salmi, Bioresour. Technol., 2015, 193, 337–344 Search PubMed.
- R. C. Narayan, J. V. Dev and G. Madras, J. Supercrit. Fluids, 2015, 101, 87–94 Search PubMed.
- H. S. Ghaziaskar, H. Eskandari and A. Daneshfar, J. Chem. Eng. Data, 2003, 48, 236–240 CrossRef CAS.
- R. A. Bourne, J. G. Stevens, J. Ke and M. Poliakoff, Chem. Commun., 2007, 4632–4634 RSC.
| This journal is © The Royal Society of Chemistry 2017 |