
Continuous flow ring-opening polymerizations
Xin
Hu†
a,
Ning
Zhu†
b,
Zheng
Fang
b and
Kai
Guo
 *bc
*bc
aCollege of Materials Science and Engineering, Nanjing Tech University, Nanjing 211800, China
bCollege of Biotechnology and Pharmaceutical Engineering, Nanjing Tech University, Nanjing 211800, China. E-mail: guok@njtech.edu.cn
cState Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, Nanjing 211800, China
First published on 9th December 2016
Abstract
Ring-opening polymerization is one of the three polymerization methodologies, together with addition polymerization and polycondensation. Poly(ester), poly(2-oxazoline), poly(phosphoester), poly(ether), poly(amino acid) and nylon 6 are mainly synthesized via ring-opening polymerizations of their corresponding cyclic monomers. To obtain well-defined and cost-effective polymers, the combination of ring-opening polymerization and microflow technology has drawn great interest from both academia and industry. This article highlighted recent progress in continuous flow ring-opening polymerizations. Lactone, lactide, 2-oxazoline and cyclic phosphate were assessed as cyclic monomers in consecutive order. The challenge and outlook for microfluidic ring-opening polymerization were also discussed.
 Xin Hu | Xin Hu was born in Xi'an in 1987. She studied Materials Science and Engineering (B.S. 2009 and Ph.D. 2014) at Xi'an Jiaotong University under the supervision of Professor Zhicheng Zhang and Professor Yuansuo Zheng. After defending her Ph.D. thesis entitled “Application of Single Electron Transfer Living Radical Polymerization (SET-LRP) in Functional Modification of PVDF-based Fluoropolymers”, she moved to Nanjing Tech University as an Assistant Professor in October 2014. Her current research interests are focused on (photo-induced) reversible deactivation radical polymerization, ring-opening polymerization, continuous flow polymerization and high performance polymeric functional materials. |
 Ning Zhu | Ning Zhu was born in Shijiazhuang in 1983. He received his Ph.D. degree in Polymer Chemistry and Physics from Zhejiang University in 2011, supervised by Professor Zhiquan Shen and Associate Professor Jun Ling. From 2010 to 2011, he studied at Université de Lille 1 as an exchange Ph.D. student under the supervision of Professor Patrice Woisel, sponsored by Erasmus Mundus. After working at Zhejiang Chemical Industry Research Institute and Xi'an University of Architecture and Technology, he joined the faculty at Nanjing Tech University (Assistant Professor 2013 and Associate Professor 2016). His current research interests include development of living/controlled polymerization methodologies, microflow technology and bio-based materials. |
 Zheng Fang | Zheng Fang was born in Nanjing in 1979. He received his M.S. degree in Medical Chemistry from China Pharmaceutical University in 2004 and Ph.D. degree in Pharmaceutical Engineering from Nanjing Tech University in 2012. He began his research work in organic synthesis as a faculty at Nanjing Tech University in 2004. He was promoted to Associate Professor in 2012. From 2015 to 2016, he took Visiting Scholar research work in organic chemistry with Professor Beining Chen at The University of Sheffield. Now, his research interests are focused on microflow chemistry, chemical engineering and bio-based materials. |
 Kai Guo | Kai Guo was born in Yancheng in 1982. He received his Ph.D. degree in Organic Chemistry from The University of Sheffield in 2008. After working as the project manager at Chempharm and as a post-doctoral researcher at The University of Sheffield, he joined the faculty at Nanjing Tech University in 2010 and was promoted to full Professor in 2011. Professor Guo's research interests are focused on flow chemistry and bio-based materials. His work has been the recipient of multiple awards including First Class Prize for Scientific and Technology Progress of China Petroleum and Chemical Industry Federation (2012), Second Class Prize for Technological Invention of State Education Ministry of China (2013 and 2016), Min-Enze Energy and Chemical Award of Chinese Academy of Engineering (2013), Industry-University-Research Innovation Award of China Industry-University-Research Institute Collaboration Association (2013) and First Class Prize for Technology Invention of China Petroleum and Chemical Industry Federation (2016). |
Introduction
Ring-opening polymerization (ROP) together with addition polymerization and polycondensation are the three polymerization methodologies for synthetic polymers.1 Many commercial polymeric materials are produced via ROPs of their corresponding cyclic monomers, such as degradable poly(ester), poly(2-oxazoline), poly(phosphoester), poly(ether), poly(amino acid) and high performance engineering plastic nylon 6.2,3 The driving force for cyclic monomer ring-opening is the relief of bond-angle strain or steric repulsions. The enthalpy change in ring-opening is negative to make the polymerization happen. Like polycondensation, ROP can construct carbon–heteroatom bonds to incorporate esters, amides and other functional groups into the polymer's main chain. Cyclic olefins can be polymerized to form carbon–carbon bonds by using metal catalysts, named ring-opening metathesis polymerization (ROMP), which is not discussed in this article.4The research interest in ROP is focused on developing novel types of catalysts.5 Remarkable advances have been made in this area to precisely control the molecular weight, molecular weight distribution and chain structure. However, scientific and engineering challenges remain for catalytic ROP.6 Another point of view might be taken to promote ROP forward besides catalyst design.
The chemical reactions conducted in traditional batch reactors (e.g. round-bottom flask) have problems with exothermic dissipation and reaction condition control. When the characteristic dimension of a reactor is miniaturized below 500 μm (or 1000 μm), it can be termed a microreactor or microfluidic device, etc., which exhibits a microscale effect in virtue of the huge volume-to-surface ratio.7–9 Microreactor-based continuous flow chemistry enables unique advantages over traditional batchwise reactions.10–14 Extremely rapid mixing and improved heat/mass transfer efficiency result in fast kinetics and high selectivity. The small internal volume (μL to mL) of a microreactor makes the reaction process safer especially for toxic or explosive reaction mixtures. The product quantities can be scaled-up by increasing the number of microreactors in parallel, which presents a convenience for industry.
In polymer chemistry, the microreactor was firstly applied in highly exothermic rapid vinyl anionic/cationic polymerization, which dated back to the pioneering work of Szwarc and co-workers in the 1960s.15 Microreactor-based continuous flow systems could offer fast mixing of reagents and strictly anhydrous conditions for ionic polymerization.16,17 Free radical polymerization was transferred from batch to the microreactor with the benefit of suppression of the Trommsdorff effect.18 Conducting living/controlled radical polymerization in continuous flow mode has become more and more popular, including atom transfer radical polymerization (ATRP),19 single electron transfer living radical polymerization (SET-LRP)20 and reversible addition fragmentation chain transfer (RAFT) polymerization.21 Cunningham, Hutchinson and co-workers reviewed Cu(0)-mediated controlled radical polymerization in a microreactor.22 Junkers and co-workers highlighted continuous photoflow synthesis of precision polymers.23 Haddleton and co-workers contributed a summary of the advances in flow SET-LRP.24 Our group evaluated continuous flow copper-mediated reversible deactivation radical polymerization.25
The first example of the combination of ROP and flow chemistry was presented by Miyazaki, Maeda and co-workers in 2005 using N-carboxyanhydrides (NCA) as cyclic monomers.26 Poly(amino acid) with a relatively narrower molecular weight distribution was prepared in a microreactor, compared with polymers obtained in a batch reactor. In 2007, Frey and co-workers reported the synthesis of hyperbranched polyglycerol in a flow microreactor.27 In the same year, Schubert and co-workers studied ROP of 2-ethyl-2-oxazoline in continuous flow mode under microwave irradiation.28 These excellent works were highlighted in several critical reviews about microfluidic polymerizations.29–31
Continuous flow ROP has been growing rapidly since 2011. The cyclic monomer scope was extended to lactone, lactide and cyclic phosphates. Enzyme immobilized microreactors, novel catalysis and photo click chemistry were combined for precise macromolecule synthesis. In this minireview, we talked about the recent important development of continuous flow opening polymerizations of lactone, lactide, 2-oxazoline and cyclic phosphate. To the best of our knowledge, there has been no flow review specifically focused on ROP. The remaining challenge and future outlook for continuous flow ring-opening polymerizations were discussed.
Continuous flow ring-opening polymerizations of lactone/lactide as the monomer
Aliphatic polyesters, such as fossil-sourced poly(ε-caprolactone) (PCL) and bio-sourced poly(lactide) (PLA), have been identified as an important class of biomaterials due to their excellent biodegradability and biocompatibility.3 The synthesis and application of these degradable polyesters have attracted growing research interest with concerns about energy and environmental issues. ROP of lactone/lactide is the main synthetic strategy to well-defined high molecular weight polyesters with advantages over polycondensation. Tremendous efforts were focused on the development of novel catalysts for the ROP process. Metal complexes, enzymes and organic molecules are considered as three catalysis systems, which showed different characteristics and mechanisms. Their catalytic polymerizations in a traditional batch reactor are limited by batch–batch variety, low level control of polymerization conditions and intermolecular/intramolecular transesterification, which brought troubles in the synthesis of high quality and cost-effective polymers.6Gross, Beers and co-workers presented an idea to improve the polyester synthesis process by transferring ROP from a batch reactor to a microreactor.32,33 Biotechnology has been growing fast to produce biofuel, bio-based chemicals and polymers as a green and sustainable protocol. Although high efficiency and selectivity are features of enzymatic transformation in vivo, industrial enzyme catalysis in vitro suffers from high cost and low efficiency. The control of the polymer's molecular weight and molecular weight distribution is also poor. Gross, Beers and co-workers fabricated an aluminium microreactor with microchannels, which were filled with Novozyme 435 (Candida antarctica lipase B (CALB)) beads (Fig. 1).32 Benzyl alcohol initiated CL polymerization in toluene was conducted in this enzyme immobilized microreactor. Compared with the batch reactor, monomers/initiators were forced to have more effective collisions with the enzyme active sites in the confined volume of the microreactor. The apparent rate constant in the microreactor (kapp = 0.027 s−1) was 27 times larger than that in the batch reactor (kapp = 0.001 s−1).33 Furthermore, in the “water saturated” microreactor, a higher molecular weight (Mn = 1000 to 2500 g mol−1) and benzyl end group fidelity (0.75 to >0.98) were achieved than those in the batch system (Mn = 500 to 1000 g mol−1, 0.2) (Fig. 2).33 The recycling number of the enzyme was about 30 under “dry” or “water saturated” conditions. This demonstrated that the green and sustainable microreactor-based continuous flow system could be extended to other enzymatic transformation processes.
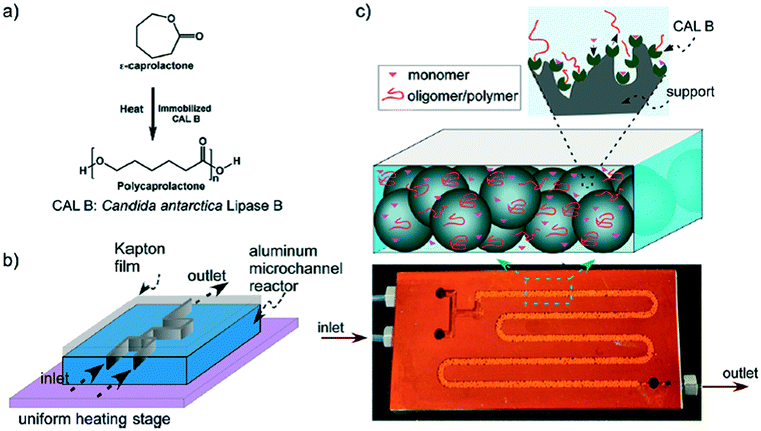 | ||
| Fig. 1 (a) Reaction scheme for ring-opening polymerization of ε-caprolactone to polycaprolactone. (b) Schematic of the microreactor setup. The microreactor was made of aluminium and covered with Kapton film using thermally cured epoxy. The microreactor was placed on a uniform heating stage for temperature control. (c) Image shows a photograph of a typical microreactor. CALB immobilized solid beads (macroporous polymethyl methacrylate) filled the channel. The polymerization reaction took place at enzyme active sites. The residence time for the reactants inside the microreactor was changed by varying the flow rate32 (reprinted with permission from ref. 32 Copyright (2011) American Chemical Society). | ||
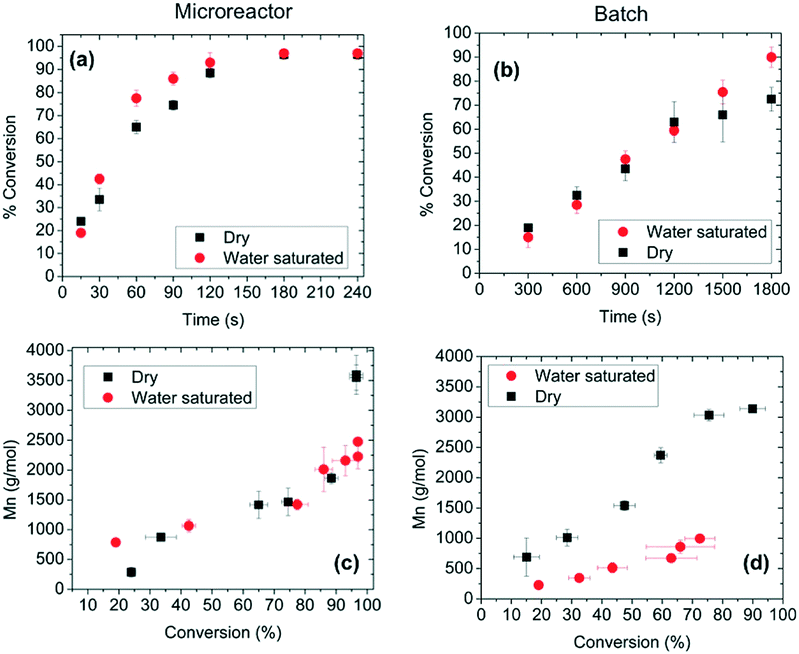 | ||
| Fig. 2 Conversion of ε-caprolactone as a function of residence or reaction time at 70 °C using benzyl alcohol as a C-terminal end functionalizing agent in the (a) microreactor and (b) batch mode. Number-average molecular mass (Mn) as a function of conversion at 70 °C in the (c) microreactor and (d) batch mode, at 70 °C, for 90 s in the microreactor and 1800 s in the batch reactor. The term “dry” represents experiments conducted using dried toluene-d8 (water content ∼0.001% w/w). The term “water saturated” denotes experiments conducted in toluene-d8 saturated with water (∼0.033% w/w). Error bars indicate one standard uncertainty based on measurements on at least three different samples33 (reprinted with permission from ref. 33 Copyright (2012) American Chemical Society). | ||
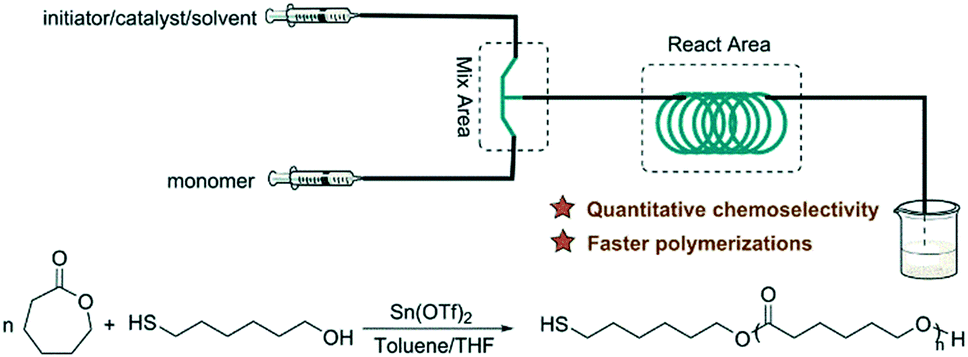
Inspired by the exciting work mentioned above, our group investigated metal- and organo-catalyzed continuous flow ROP. Stannous(II) is a typical metal catalyst for cyclic ester polymerization, which is authorized by the FDA and used in commercial polyester production in industry.34 Stannous(II) trifluoromethane sulfonate (Sn(OTf)2) was chosen for model investigation due to its commercial availability, simplicity of handling and tolerance to moisture. We established a simple PTFE tubular microreactor with a T-type mixer (Fig. 3).35 Under the same reaction conditions, CL polymerization with benzyl alcohol as the initiator proceeded faster in the microreactor than in batch mode (90.1% vs. 81.8% monomer conversion for 40 min). In virtue of intensified mixing and mass/heat transfer efficiency, inter/intramolecular transesterification was suppressed, which resulted in smaller polydispersities in the microreactor for different target degrees of polymerization (DPtarget) (PDI = 1.15 vs. 1.30 (DPtarget = 10), PDI = 1.24 vs. 1.32 (DPtarget = 30)) (Fig. 4).35
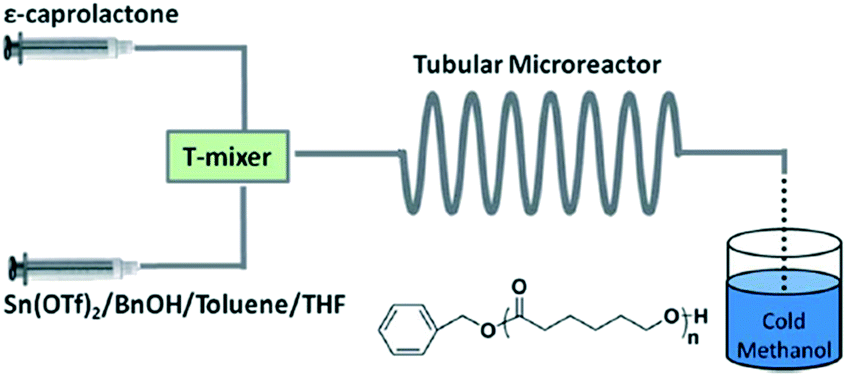 | ||
| Fig. 3 Sn(OTf)2 catalyzed continuous flow ring-opening polymerization of ε-caprolactone in toluene/THF in a PTFE tubular microreactor with a T-type mixer35 (reproduced from ref. 35 with permission from The Royal Society of Chemistry). | ||
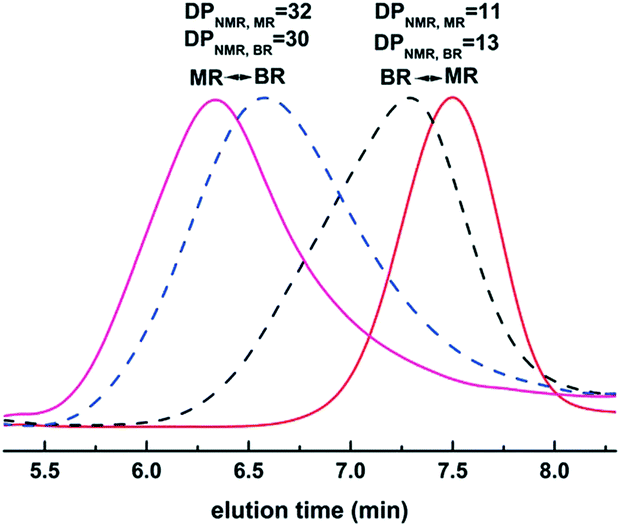 | ||
| Fig. 4 SEC of PCL with DPtarget = 10 at 80 °C (40 min in the microreactor and 80 min in the batch reactor) and DPtarget = 30 at 110 °C (80 min in the microreactor and batch reactor)35 (reproduced from ref. 35 with permission from The Royal Society of Chemistry). | ||
Organocatalyzed ROP has made extraordinary advances in preparing metal-free polymers for biomedical and microelectronic applications.36 Various kinds of organic molecules have been selected to perform polymerizations with different proposed mechanisms. Commercially available 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) is a highly active organic catalyst for ROP of lactone, lactide, carbonate and carbosiloxane with a “bifunctional activation of monomer and initiator/chain end” mechanism.37 However, it could also catalyze transesterification, which leads to an undesired increase in the molecular weight distribution upon full monomer conversion. Zuilhof, Wennekes and co-workers optimized the reaction temperature and residence time of TBD catalysed continuous flow ROP of L-lactide (Fig. 5).38 More than 95% L-lactide conversion and narrow polydispersity (PDI < 1.3) were obtained under −10 to 30 °C at residence times as low as 2 s with moderate catalyst loading (0.25–1.2%). Post-functionalization of PLA was carried out via click chemistry by using a functional initiator, which allowed the expansion of the scope of flow-based polymerizations for biomedical and other applications.
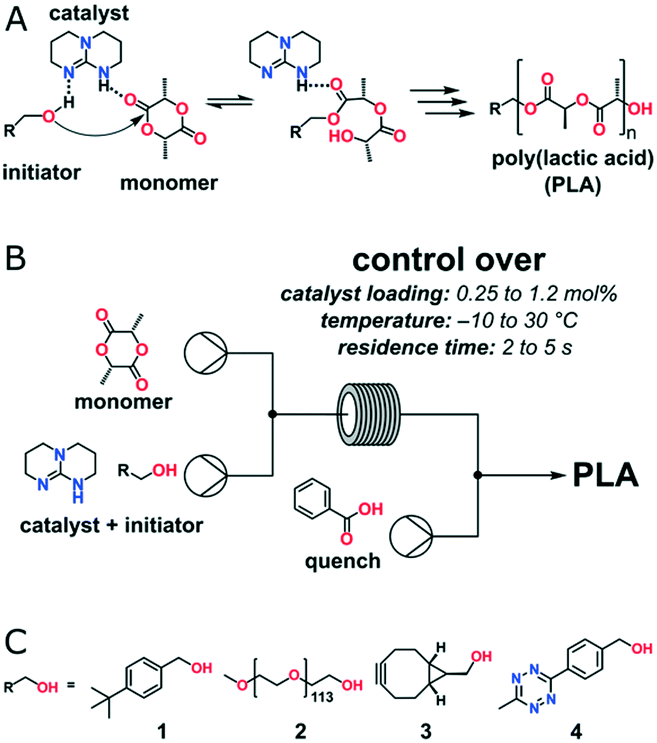 | ||
| Fig. 5 (A) TBD-catalyst mode of action in the ring-opening polymerization of L-lactide: hydrogen bonding increases the nucleophilicity of the alcohol initiator. (B) The continuous flow organo-catalytic polymerization of L-lactide is performed in a commercially available microreactor setup using a 1 μL internal volume glass microreactor chip. (C) Alcohol initiators: 4-tert-butylbenzyl alcohol 1, PEG macroinitiator 2, exo-BCN-alcohol 3, and tetrazine 438 (reprinted with permission from ref. 38 Copyright (2016) American Chemical Society). | ||
Simultaneously, our group systematically studied the kinetics of TBD catalyzed continuous flow polymerization of CL and δ-valerolactone (VL) (Fig. 6).39 The molecular weights of PCL (Mn,NMR = 2290 to 3480 g mol−1) and PVL (Mn,NMR = 2210 to 9120 g mol−1) were obtained by modulating the flow rates of the PTFE tubular microreactor. Apparent rate constants in the microreactor were almost two times bigger than those in the batch reactor for both CL and VL at different DPtarget. By assembling an integrated-microreactor system, easier and more efficient block copolymerizations were successfully conducted by sequential introduction of monomers to prepare well-defined PVL–block-PCL and PCL–block-PVL (Fig. 7). This provided the potential application of synthesizing multi-block or sequence-controlled macromolecules.
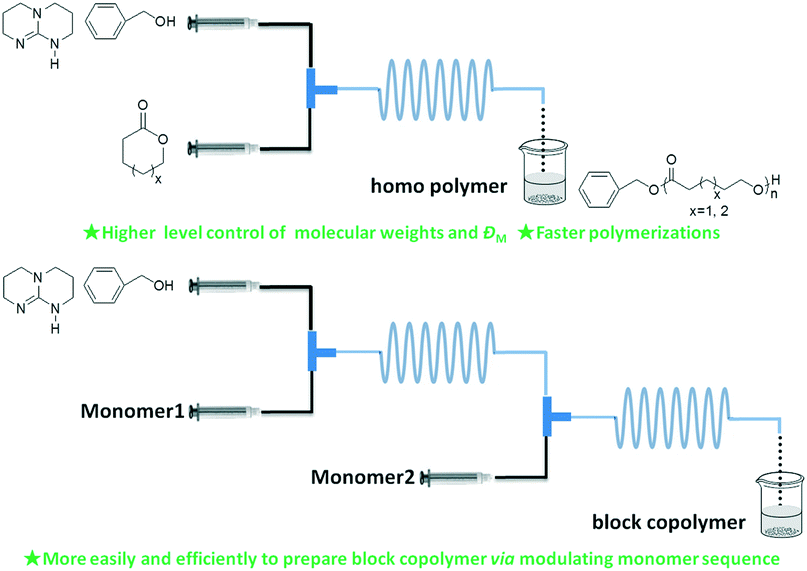 | ||
| Fig. 6 TBD catalyzed ring-opening polymerization of ε-caprolactone and δ-valerolactone to homo- and block-polylactones in a microreactor39 (reproduced from ref. 39 with permission from Elsevier). | ||
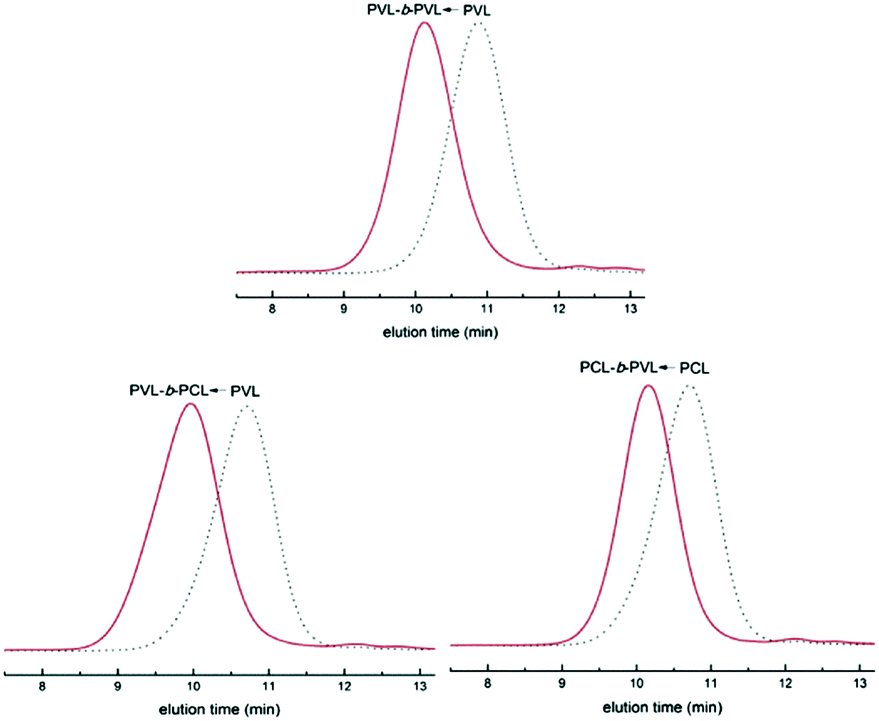 | ||
| Fig. 7 SEC traces of PVL–b-PVL, PVL–b-PCL and PCL–b-PVL prepared in an integrated microreactor39 (reproduced from ref. 39 with permission from Elsevier). | ||
Chemoselective reaction has been a fashionable research topic in organic and polymeric chemistry. Protecting-group-free synthesis has attracted significant attention as an ideal and green sustainable method.40 The thiol group exhibited special chemical and physical activities in click chemistry, forming sulfur–metal and disulfide bonds. Besides, it could also initiate ring-opening polymerization like the hydroxyl group. To obtain thiol-functionalized polyesters, tedious protecting/deprotecting steps for the mercapto group should be involved during polymerization in the presence of thiol-alcohol as the initiator.41 Research work has been done to explore chemoselective catalysts toward hydroxyl and mercapto. Candida antarctica Lipase B (CALB),42 lipase from Candida sp. 99–125,43 Sn(OTf)2,44 and rare earth phenolates45,46 were reported to directly catalyze ROP without protecting thiol. The microreactor could enable chemical reactions that cannot be done in batch. A protecting-group-free method for ketone reactions in the presence of organolithium species was successfully achieved by employing flow chemistry.40 Our group combined the published catalyst Sn(OTf)2 and a microreactor to develop a continuous flow protecting-group-free synthesis approach to thiol-terminated PCL (Mn = 1270 to 4580 g mol−1, PDI = 1.11 to 1.35) (Fig. 8).47 Due to the improved mixing and mass/heat transfer efficiency, the reaction time to reach over 95% CL conversion was decreased from 24 h (batch reactor) to 5–8 h (microreactor). With the benefit of flow mode and no back-mixing characteristic of the tubular reactor, the competitive thiol-related side reactions were strongly supressed and quantitative thiol fidelities (95–99%) were achieved. This might find potential application in chemoselective polymerization based on microreactor technology.
 | ||
| Fig. 8 Continuous flow chemoselective ring-opening polymerization of ε-caprolactone in a PTFE tubular reactor to thiol-terminated poly(ε-caprolactone)47 (reproduced from ref. 47 with permission from Elsevier). | ||
Continuous flow ring-opening polymerizations of 2-oxazoline as the monomer
Poly(2-oxazoline) (POx) has been paid much attention for their biocompatibility, tunable hydrophilic/hydrophobic properties and diverse stimuli-responsiveness.48 Poly(2-oxazoline)s with short side chains, such as poly(2-ethyl-2-oxazoline) (EtOx) and poly(2-n-propyl-2-oxazoline) (nPropOx), are considered as promising alternatives to poly(ethylene glycol) (PEG). To reduce the long-term polymerization time and overcome the one-at-a-time nature of polymerization in single microwave vials, Schubert and co-workers made use of continuous-flow microwave reactors to conduct cationic ROP of 2-ethyl-2-oxazoline.28 Broader molecular weight distributions of the resulting poly(EtOx) were observed in continuous flow mode. It was supposed to be attributed to the residence time distributions which were determined by using methyl orange as the flow marker.Hoogenboom, Junkers and co-workers evaluated continuous flow cationic ROP of 2-ethyl-2-oxazoline (EtOx) and 2-n-propyl-2-oxazoline (nPropOx) with regard to reaction temperature.49 Fast homopolymerizations with full monomer conversions were observed after 12 min 30 s at 140 °C, 5 min at 160 °C and 2 min at 180 °C for both monomers. Moreover, diblock and triblock copolymers were more easily and efficiently synthesized by using a microfluidic reactor cascade without a polymer isolation step (Fig. 9).49 The introduction of any moisture and intensive labour consuming process during multistep comonomer addition in batch mode could be avoided by the utility of microflow technology. This indicated the high value and promising future of flow chemistry in poly(2-oxazoline)-based polymer synthesis.
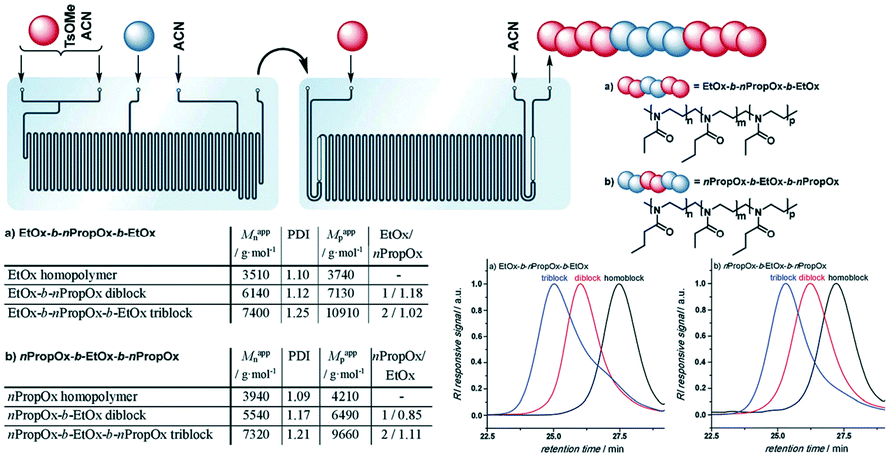 | ||
| Fig. 9 Schematic representation of the microfluidic cascade consisting of a two stage 15 μL and 19.5 μL microreactor. Injection of a pure monomer (in a 1/1 ratio to the first monomer) via the additional inlet in the 15 μL reactor enables the formation of diblock copolymers, while the injection of a monomer in the 19.5 μL reactor leads to triblock copolymers. (a) EtOx–b-nPropOx–b-EtOx: 5 min + 6 min 48 s + 6 min 58 s; (b) nPropOx–b-EtOx–b-nPropOx: 5 min + 7 min 09 s + 6 min 58 s49 (reproduced from ref. 49 with permission from The Royal Society of Chemistry). | ||
Continuous flow ring-opening polymerizations of cyclic phosphate as the monomer
Poly(phosphoester) is biocompatible and hemocompatible with structural similarities to nucleic acids.50 The side chains could be designed on request with different solubilities and properties. Although poly(phosphoester) could be prepared via polycondensation and transesterification, metal- and organo-catalytic anionic ROP is the common synthesis approach to polymers with controlled molecular weight and narrow molecular weight distribution. Currently, the stringent reaction conditions, considerable batch–batch variations and the difficulties in scaling up this process need to be overcome.Lecomte, Junkers and co-workers conducted anionic ROP of cyclic phosphates to yield poly(phosphoester) under continuous flow conditions.51 Almost full conversion of 2-isobutyoxy-2-oxo-1,3,2-dioxaphospholane (iBP) was achieved within 10 min or 3 min at 40 °C catalyzed by TBD and 1,8-diazobicyclo[5.4.0]undec-7-ene (DBU)/(thiourea) (TU), respectively. 2-Butenoxy-2-oxo-1,3,2-dioxaphospholane (BP) was polymerized under optimized conditions to obtain polymers with an alkene functionality. Thiol-ene post-modification proceeded with high efficiency under UV. Further, they coupled thermally-activated anionic polymerization and UV-induced thiol-ene post-modification directly in a sequence-connected microreactor system (Fig. 10).51 Functionalized polymers were straightforwardly obtained without isolation. This microreactor system can be used to prepare novel functional poly(phosphoester)-based materials with various properties.
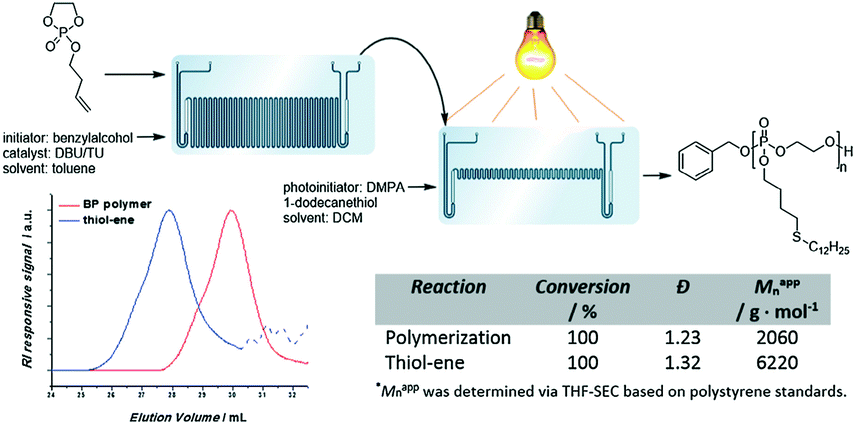 | ||
| Fig. 10 Schematic representation of the microfluidic cascade consisting of a two stage 19.5 μL and 5 μL microreactor. An alkene-functionalized BP polymer is formed in the first reactor, after which the polymer is directly functionalized via a UV-induced thiol-ene reaction51 (reproduced from ref. 51 with permission from Elsevier). | ||
Conclusions
Microreactor-based continuous flow polymerization has exhibited numerous advantages over traditional batchwise reactions. Excellent heat and mass transfer, efficient mixing and short diffusion paths enable polymerizations with faster rates, narrower molecular weight distribution, higher yields and selectivity. High resolution control of the reaction conditions, ease of scaling up and a safe polymerization process are also achieved by using flow chemistry.Compared with anionic/cationic polymerization, free radical polymerization and living/controlled radical polymerization, continuous flow ring-opening polymerization is a young yet rapidly growing research area. Since 2011, the cyclic monomer scope has been extended to ε-caprolactone, δ-valerolactone, L-lactide, 2-ethyl-oxazoline, 2-n-propyl-oxazoline, 2-isobutyoxy-2-oxo-1,3,2-dioxaphospholane and 2-butenoxy-2-oxo-1,3,2-dioxaphospholane. Enzyme immobilized microchannel reactors and tubular reactors were fabricated to perform continuous flow ring-opening polymerizations associated with click chemistry. Although tremendous advances were made by transferring ROP from batch into a microreactor, the potential of continuous flow ROP is not yet completely explored. Attention should be paid to the viscosity increase in the flow process. The rector plug could be observed with the aim of synthesizing high molecular weight polymers. The scientific research and industrial application of microreactor-based ROP still have a long way. More cyclic monomers need to be evaluated under a flow system, including thioether, thiolactone, disulphide, anhydride, carbonate, silicone and phosphazene. Besides homopolymer and di- or tri-block copolymer synthesis, multiblock and sequence-controlled polymer synthesis might be conducted in continuous flow mode. It is believed that light triggered ROP will be developed forward by both catalyst design and utility of microreactor technology in the future.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21504039, 21604037 and 21522604) and the Jiangsu National Synergetic Innovation Centre for Advanced Materials (SICAM).Notes and references
- O. Nuyken and S. D. Pask, Polymers, 2013, 5, 361–403 CrossRef.
- N. Zhu, H. Gong, W. Han, W. Zeng, H. Wang, Z. Fang, X. Li, K. Zhang, Z. Li and K. Guo, Chin. Chem. Lett., 2015, 26, 1389–1392 CrossRef CAS.
- O. Dechy-cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- S. M. Guillaume, E. Kirillov, Y. Sarazin and J. Carpentier, Chem. – Eur. J., 2015, 21, 7988–8003 CrossRef CAS PubMed.
- K. S. Elvira, X. C. Solvas, R. C. R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef CAS PubMed.
- J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC.
- J. Poh, D. L. Browne and S. V. Ley, React. Chem. Eng., 2016, 1, 101–105 CAS.
- B. J. Reizman and K. F. Jensen, Acc. Chem. Res., 2016, 49, 1786–1796 CrossRef CAS PubMed.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61–67 CrossRef CAS PubMed.
- H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noël, Chem. Soc. Rev., 2016, 45, 83–117 RSC.
- I. M. Mándity, S. B. Ötvös and F. Fülöp, ChemistryOpen, 2015, 4, 212–223 CrossRef PubMed.
- I. M. Mándity, S. B. Ötvös, G. Szőlősi and F. Fülöp, Chem. Rec., 2016, 16, 1018–1033 CrossRef PubMed.
- C. Geacintov, J. Smid and M. Szwarc, J. Am. Chem. Soc., 1962, 84, 2508–2514 CrossRef CAS.
- A. Nagaki, A. Miyazaki and J. Yoshida, Macromolecules, 2010, 43, 8424–8429 CrossRef CAS.
- A. Nagaki, K. Kawamura, S. Suga, T. Ando, M. Sawamoto and J. Yoshida, J. Am. Chem. Soc., 2004, 126, 14702–14703 CrossRef CAS PubMed.
- T. Iwasaki and J. Yoshida, Macromolecules, 2005, 38, 1159–1163 CrossRef CAS.
- D. Parida, C. A. Serra, D. K. Garg, Y. Hoarau, F. Bally, R. Muller and M. Bouquey, Macromolecules, 2014, 47, 3282–3287 CrossRef CAS.
- N. Zhu, X. Hu, Y. Zhang, K. Zhang, Z. Li and K. Guo, Polym. Chem., 2016, 7, 474–480 RSC.
- J. Vandenbergh, T. Ogawa and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2366–2374 CrossRef CAS.
- N. Chan, M. F. Cunningham and R. A. Hutchinson, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3081–3096 CrossRef CAS.
- T. Junkers and B. Wenn, React. Chem. Eng., 2016, 1, 60–64 CAS.
- A. Anastasaki, V. Nikolaou, A. Simula, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2016, 116, 835–877 CrossRef CAS PubMed.
- X. Hu, N. Zhu, Z. Fang, Z. Li and K. Guo, Eur. Polym. J., 2016, 80, 177–185 CrossRef CAS.
- T. Honda, M. Miyazaki, H. Nakamura and H. Maeda, Lab Chip, 2005, 5, 812–818 RSC.
- D. Wilms, J. Nieberle, J. Klos, H. Löwe and H. Frey, Chem. Eng. Technol., 2007, 30, 1519–1524 CrossRef CAS.
- R. M. Paulus, T. Erdmenger, C. Remzi Becer, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2007, 28, 484–491 CrossRef CAS.
- C. Tonhauser, A. Natalello, H. Löwe and H. Frey, Macromolecules, 2012, 45, 9551–9570 CrossRef CAS.
- A. Nagaki and J. Yoshida, Adv. Polym. Sci., 2013, 259, 1–50 CAS.
- J. L. Steinbacher and D. T. McQuade, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6505–6533 CrossRef CAS.
- S. Kundu, A. S. Bhangale, W. E. Wallace, K. M. Flynn, C. M. Guttman, R. A. Gross and K. L. Beers, J. Am. Chem. Soc., 2011, 133, 6006–6011 CrossRef CAS PubMed.
- A. S. Bhangle, K. L. Beers and R. A. Gross, Macromolecules, 2012, 45, 7000–7008 CrossRef.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 689–695 CrossRef CAS.
- N. Zhu, Z. Zhang, W. Feng, Y. Zeng, Z. Li, Z. Fang, K. Zhang, Z. Li and K. Guo, RSC Adv., 2015, 5, 31554–31557 RSC.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- S. A. Van den Berg, H. Zuilhof and T. Wennekes, Macromolecules, 2016, 49, 2054–2062 CrossRef CAS.
- N. Zhu, W. Feng, X. Hu, Z. Zhang, Z. Fang, K. Zhang, Z. Li and K. Guo, Polymer, 2016, 84, 391–397 CrossRef CAS.
- H. Kim, A. Nagaki and J. Yoshida, Nat. Commun., 2011, 2, 264 CrossRef PubMed.
- M. Trollsås, C. J. Hawker and J. L. Hedrick, Macromolecules, 1998, 31, 5960–5963 CrossRef.
- C. Hedfors, E. östmark, E. Malmström, K. Hult and M. Martinelle, Macromolecules, 2005, 38, 647–649 CrossRef CAS.
- N. Zhu, Z. Zhang, W. He, X. Geng, Z. Fang, X. Li, Z. Li and K. Guo, Chin. Chem. Lett., 2015, 26, 361–364 CrossRef CAS.
- N. Xu, R. Wang, F. Du and Z. Li, Chem. J. Chin. Univ., 2007, 28, 1791–1795 CAS.
- N. Zhu, J. Ling, Y. H. Zhu, W. Sun and Z. Shen, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4366–4369 CrossRef CAS.
- N. Zhu, W. Feng, Z. Zhang, Z. Fang, Z. Li and K. Guo, Polymer, 2015, 80, 88–94 CrossRef CAS.
- N. Zhu, Y. Liu, W. Feng, W. Huang, Z. Zhang, X. Hu, Z. Fang, Z. Li and K. Guo, Eur. Polym. J., 2016, 80, 234–239 CrossRef CAS.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed.
- E. Baeten, B. Verbraeken, R. Hoogenboom and T. Junkers, Chem. Commun., 2015, 51, 11701–11704 RSC.
- B. Clément, B. Grignard, L. Koole, C. Jérôme and P. Lecomte, Metal-free strategies for the synthesis of functional and well-defined polyphosphoesters, Macromolecules, 2012, 45, 4476–4486 CrossRef.
- E. Baeten, S. Vanslambrouck, C. Jérôme, P. Lecomte and T. Junkers, Eur. Polym. J., 2016, 80, 208–218 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |