Methanation of residual syngas after LPG synthesis: identifying the main effects on catalytic performance with Plackett–Burman screening design†
Received
31st March 2016
, Accepted 30th June 2016
First published on 20th July 2016
Abstract
Methanation of residual syngas as a downstream process after liquefied petroleum gas (LPG) synthesis is considered to be an attractive alternative to conventional gas separation. Seven intensively studied factors in catalyst development (ratio of Fe/Ni, metal loading, selection of Al2O3 or SiO2 as oxidic support, addition of promoters such as potassium and/or MgO, heat dissipation by diluting with inert material and two common preparation techniques (impregnation and precipitation)) were selected within a Plackett–Burman screening design and rated towards their impact on catalytic performance such as carbon monoxide and hydrocarbon conversion as well as methane and carbon dioxide selectivity. Detailed characterization of fresh and used catalysts has been carried out applying N2-physisorption, X-ray diffraction (XRD), inductively coupled plasma atomic emission spectroscopy (ICP-AES), temperature-programmed reduction (H2-TPR), and transmission electron microscopy with energy-dispersive spectroscopy (TEM/EDS) in order to confirm the results. Finally, suggestions on the preferred levels of these factors are made for development of advanced catalysts.
Introduction
The energy and chemistry sectors demand tailored catalysts for efficient processes. Industry in coal-rich countries, for example, considers methanation (eqn (1) and (2)) as a means to produce substitute natural gas (SNG) from coal for feeding into their national gas grid in order to become independent from imports.1 Methanation (Sabatier reaction) on its own has been known since the 20th century2 and has been a research focus for decades.3 Methanation is already effectively applied in removal of CO from hydrogen-rich gas streams4–7 and in production of synthetic natural gas (SNG).3,8,9| | CO(g) + 3H2(g) ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) CH4(g) + H2O(g) ΔH0 = −206 kJ mol−1 CH4(g) + H2O(g) ΔH0 = −206 kJ mol−1 | (1) |
| | | CO2 (g) + 4H2(g) CH4(g) + 2H2O(g) ΔH0 = −165 kJ mol−1 | (2) |
In such a concept, SNG will be usually blended with small quantities of liquefied petroleum gas (LPG) to meet the calorimetric demands for the gas grid. However, in LPG synthesis, residual syngas remains after reaction. Removing excess CO and CO2via methanation enables direct use without further purification and appears to be an economically attractive alternative compared to conventional gas separation. However, the development of solid catalysts for methanation of residual syngas in the presence of LPG components is still a demanding task since the literature is scarce. From a thermodynamic point of view, traces of light hydrocarbons in the syngas feed will change the picture distinctly.10 Conversion of CO will not be affected while methane selectivity decreases significantly. Especially, the risk of carbon formation rises drastically. Even traces of hydrocarbons (7%) in the feed increase the carbon yield to ≈13% at 300 °C and ≈47% at 600 °C.3,10 Therefore, the major challenge to overcome relates to cracking of LPG components taking place along with CO hydrogenation, which in turn causes catalyst deactivation by coking. For nickel catalysts, deactivation also takes place at low temperatures (< ca. 190 °C) due to formation of carbonyls.11,12 Therefore, a moderate temperature has to be selected.
The development of a catalyst is a time-consuming process and, unfortunately, time is often a limited resource. Identifying the impact on catalytic performance for a wide range of factors by a one-factor-at-a-time approach or educated guesses is a rather slow process. Design of Experiments (DoE) provides several screening methodologies known to be more efficient. These methodologies enable identifying relationships between multiple inputs and certain outputs at once. In addition, a factor's influence on certain responses is not only identified but also ranked according to its impact on these responses. Therefore, an appropriate screening method can provide detailed knowledge about a system, while saving time and experimental expense.
In this work, we applied a Plackett–Burman design13 in catalyst development for methanation of residual syngas from LPG synthesis. The identification of main factors (variables) affecting catalyst performance is demonstrated as well. Additionally, suggestions for the desired variable levels are made. The properties of the evaluated catalysts, denoted as responses according to the Plackett–Burman terminology, are rated in terms of carbon monoxide and propane conversion (propane is used as a model LPG compound), as well as methane and carbon dioxide selectivity. Moreover, detailed characterization of fresh and used catalysts has been carried out applying N2-physisorption, X-ray diffraction (XRD), inductively coupled plasma atomic emission spectroscopy (ICP-AES), temperature-programmed reduction (H2-TPR), and transmission electron microscopy with energy-dispersive spectroscopy (TEM/EDS) to support the results.
Experimental
Selection of variables/factors
The catalytic activity of heterogeneous catalysts depends on plenty of factors. Based on a thorough methanation literature survey,3,14–18 we selected seven factors, i.e. variables expected to exhibit the strongest effects on the final catalyst performance (Table 1): molar ratio of active metals (A), amount of metal loading (B), oxidic supports with different degrees of acidity/basicity (C), use of support promoter (D) and alkali dopant (E), dilution with inert material (F) as well as commonly used preparation techniques (G). A more detailed description of the individual factors is given below.
Table 1 Assigning the seven coded factors (A–G) each with two levels (+/−) to either numerical or categorical values
|
|
Factor |
Level |
| − |
+ |
| A |
Molar ratio of Fe/Ni |
0/1 |
1/1 |
| B |
Metal loading |
5 wt% |
15 wt% |
| C |
Support |
Al2O3 |
SiO2 |
| D |
MgO |
0 wt% |
10 wt% |
| E |
Potassium |
0 wt% |
5 wt% |
| F |
Quartz/catalyst |
0/1 |
1/1 |
| G |
Preparation method |
Impregnation |
Precipitation |
Catalytic systems for sole CO methanation are based on VIIIB metals with various oxidic supports.19–21 The most important ones are Ru, Ni, Co, Fe and Mo,15 wherein Ni is the most studied system.16 Recently, bimetallic Ni–Fe alloys were identified to be more active and less expensive than traditional Ni-based catalysts.22 Therefore, with respect to application in industry, the behavior of pure nickel and Ni/Fe alloys in the presence of hydrocarbons is interesting since iron (and nickel) can also produce hydrocarbons known from Fischer–Tropsch chemistry.23 The upper limit for the metal loading has to be restricted to 15–20 wt% to allow preparation via incipient wetness impregnation. In contrast, low loadings (e.g. 5 wt%) are considered to have better dispersion. For the influence of the support, SiO2 was selected as an acidic support and Al2O3 as an amphoteric support, since both are frequently applied. In addition, MgO is a well-known promoter for increasing the basicity and enhancing the resistance against carbon deposition.24 Similarly, potassium can suppress coke formation and prevents sintering of the active metal.25 Enhanced heat dissipation is favorable to avoid pronounced carbon formation.10 Therefore, dilution with an inert material (quartz) is considered. Finally, two commonly applied preparation techniques (impregnation and precipitation) are chosen, because large quantities of catalyst can be produced in industry.
The Plackett–Burman screening approach used for these seven factors with two levels each is a fractional factorial design resulting in eight different catalysts as presented in Tables 2 and 3. What makes it special is that the experimental expenses were substantially decreased from a total of 128 combinations as required by a full factorial design to only eight. Based on the reduced effort, the entire set was replicated to crosscheck for variations and reproducibility.
Table 2 Plackett–Burman 8-run matrix for seven factors (A–G) with two levels (+/−) resulting in eight catalysts (1–8)
|
|
|
Factors |
|
|
N = 8 |
A |
B |
C |
D |
E |
F |
G |
| Catalysts |
1
|
+ |
+ |
+ |
− |
+ |
− |
− |
|
2
|
− |
+ |
+ |
+ |
− |
+ |
− |
|
3
|
− |
− |
+ |
+ |
+ |
− |
+ |
|
4
|
+ |
− |
− |
+ |
+ |
+ |
− |
|
5
|
− |
+ |
− |
− |
+ |
+ |
+ |
|
6
|
+ |
− |
+ |
− |
− |
+ |
+ |
|
7
|
+ |
+ |
− |
+ |
− |
− |
+ |
|
8
|
− |
− |
− |
− |
− |
− |
− |
Table 3 Summary of the eight prepared catalysts according to Plackett–Burman design with seven different input factors each with two levels
| Catalyst |
Fe/Ni (mol/mol) |
Metal loading (wt%) |
Support |
MgO promoter (wt% rel. to support) |
K wt% of final catalyst |
Quartz/catalyst |
Preparation methoda |
|
IWI: incipient wetness impregnation, DP: deposition precipitation, P: (co)-precipitation.
|
|
1
|
1 |
15 |
SiO2 |
0 |
5 |
0 |
IWI |
|
2
|
0 |
15 |
SiO2 |
10 |
0 |
1 |
IWI |
|
3
|
0 |
5 |
SiO2 |
10 |
5 |
0 |
DP |
|
4
|
1 |
5 |
Al2O3 |
10 |
5 |
1 |
IWI |
|
5
|
0 |
15 |
Al2O3 |
0 |
5 |
1 |
P |
|
6
|
1 |
5 |
SiO2 |
0 |
0 |
1 |
DP |
|
7
|
1 |
15 |
Al2O3 |
10 |
0 |
0 |
P |
|
8
|
0 |
5 |
Al2O3 |
0 |
0 |
0 |
IWI |
Catalyst preparation
Due to the different chemical nature of alumina and silica, two modified precipitation approaches were used for each of them: (co)precipitation (P) for alumina and deposition precipitation (DP) for silica. Incipient wetness impregnation (IWI) was the impregnation method selected for this study. All prepared catalysts were dried at 120 °C for at least 2 h and subsequently calcined in air at 400 °C for 5 h with a heating rate of 4.0 °C min−1. Detailed descriptions of the different preparation methods are provided below. When required, potassium was added later to the catalyst precursors in an additional step by IWI with aqueous potassium carbonate solution. All chemicals and solvents were used as commercially obtained without further purification. A full list of the chemicals with their purities can be found in the ESI† (S1).
Incipient wetness impregnation.
Respective amounts of metal nitrates, Ni(NO3)2·6H2O, Mg(NO3)2·6H2O and Fe(NO3)3·9H2O, were dissolved in distilled water and added dropwise at room temperature to the agitated solid supports up to incipient wetness.26 In the case of silica, a commercial NORPRO catalyst carrier (SS61137, Saint Gobain) was crushed to 0.8–1.0 mm fractions. For alumina, 1.0 mm spheres from Sasol (γ-alumina) were used. The impregnated support was dried for 3 h at 120 °C. The impregnation and drying steps were repeated when necessary until the entire nitrate solution was absorbed by the support.
(Co)precipitation.
All metal nitrates, Al(NO3)3·9H2O, Mg(NO3)2·6H2O, Fe(NO3)3·9H2O and Ni(NO3)2·6H2O, were dissolved in distilled water. The pH was slowly increased with 12 M NaOH solution up to pH 10.27 The precipitated solid was filtered and washed with distilled water until the pH of the filtrate became 7. Finally, the calcined powder was pelletized at 40 bar for 6 min, crushed and sieved to 0.8–1.0 mm fractions.
Deposition precipitation
For deposition precipitation, chemical hydrolysis of urea was selected as a method for slowly increasing the reactant solution pH.27 In principle, the precipitation induced by hydrolysis of urea is similar to precipitation performed with alkali bases. However, due to the high reactivity of silica with alkali bases, the latter cannot be used as precipitating agents. In the first step, urea decomposes to ammonium cyanate. The subsequent hydrolysis of the cyanate, releasing hydroxide ions, can be temperature-controlled. The respective amounts of metal nitrates, Fe(NO3)3·9H2O and Ni(NO3)2·6H2O, were dissolved in distilled water and the finely crushed NORPRO silica catalyst carrier and MgO were added to the mixture. The mixture was agitated and heated to 80 °C. The deposition precipitation was initiated by adding urea in 4-fold excess. After 24 h under reflux, the reaction was stopped. The mixture was filtered and washed several times with distilled water to neutral pH of the filtrate. The calcined powder was pelletized at 40 bar for 6 min and crushed to 0.8–1.0 mm fractions.
Characterization
The prepared catalysts were extensively characterized by N2-physisorption, XRD, ICP-AES, H2-TPR, and electron microscopy in transmission and scanning transmission modes (TEM, STEM). The microscopy analyses were combined with elemental composition studies performed by EDS. The detailed descriptions of all analytical methods used are provided in the ESI† (Section S2).
CO methanation
Catalytic experiments were conducted in a tubular fixed bed reactor setup. A more detailed description can be found in the ESI† (Section S3). Catalytic tests were performed at ambient pressure, a gas inlet temperature of 290 °C and a GHSV of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 h−1. The feed composition was set to CO:H2:C3H8 = 1.0:3.0:2.4 with 10 vol% N2 as an internal standard to account for volume contraction during reaction. The reactor output was monitored by using an on-line gas chromatograph. A blank experiment, used as a control experiment, showed no activity of the setup. All catalysts were reduced in situ prior to reaction for 2 h in 50 mL min−1 H2 at 600 °C selected based on TPR measurements.
400 h−1. The feed composition was set to CO:H2:C3H8 = 1.0:3.0:2.4 with 10 vol% N2 as an internal standard to account for volume contraction during reaction. The reactor output was monitored by using an on-line gas chromatograph. A blank experiment, used as a control experiment, showed no activity of the setup. All catalysts were reduced in situ prior to reaction for 2 h in 50 mL min−1 H2 at 600 °C selected based on TPR measurements.
Results and discussion
Physicochemical properties of the prepared catalysts
N2-physisorption.
The prepared catalysts were analyzed after calcination but prior to reduction. Both sets showed pairwise comparable sorption patterns, most of them of type IV with H2 or H4 hysteresis loops (Fig. 1), with the exception of catalyst 1, which showed no intrinsic pore system (type II).
 |
| | Fig. 1 N2-physisorption isotherms for the first catalyst set. Diagrams are grouped for silica-based (left) and alumina-based catalysts (right). For the sake of clarity, the second set is not presented (no significant variations to the first set were observed). | |
Taking a look at silica-based catalysts (Table 4), a higher specific surface area of catalysts prepared by precipitation compared to catalysts prepared by impregnation becomes apparent. In the case of subsequent impregnation of catalysts 1 and 3 with potassium, the specific surface area as well as pore volume decrease potentially due to pore blocking. For catalyst 1, this effect is very pronounced; however, also alumina-based systems show a similar trend.
Table 4 Summary of specific surface area (BET) and pore volume based on N2-physisorption. Both separately prepared catalyst sets are very comparable
| Catalyst |
Preparation methoda |
Spec. surface areab (m2 g−1) |
Pore volumec (cm3 g−1) |
| Set 1 |
Set 2 |
Set 1 |
Set 2 |
|
IWI: incipient wetness impregnation, DP: deposition precipitation, P: precipitation.
Multipoint BET for 7 points in the relative pressure range p/p0 of 0.05–0.20.
Pore volumes at the highest measured relative pressure p/p0 around 0.95.
|
| Silica support |
129 |
0.24 |
|
1
|
IWI |
25 |
28 |
0.07 |
0.11 |
|
2
|
IWI |
88 |
87 |
0.29 |
0.29 |
|
3
|
DP |
90 |
91 |
0.26 |
0.25 |
|
6
|
DP |
166 |
164 |
0.47 |
0.47 |
| Alumina support |
153 |
0.33 |
|
4
|
IWI |
109 |
111 |
0.29 |
0.29 |
|
8
|
IWI |
152 |
160 |
0.46 |
0.46 |
|
5
|
P |
224 |
207 |
0.22 |
0.24 |
|
7
|
P |
281 |
284 |
0.37 |
0.35 |
Catalyst composition
The elemental composition of the prepared catalysts was analyzed by ICP-AES. Most of the mass concentrations are in close proximity to the theoretical values (ESI† Section S4). Unfortunately, the amount of potassium was too low for reliable quantification. Nevertheless, the Fe:Ni = 1:1 ratio as well as the metal loading, 5 wt% and 15 wt%, respectively, could be confirmed.
Reduction of calcined catalysts
The prepared and calcined catalysts were analyzed by H2-TPR as illustrated in Fig. 2. The materials exhibit various reduction steps and overlaps of different reduction peaks attributable to different species. Due to the complex nature of the individual materials, no further assignments of the peaks were attempted. Nevertheless, the TPR results are suggestive of the final reduction temperature (in situ) before the catalytic tests. A reduction temperature of 600 °C was selected as a compromise to obtain enough reduced species, on one side, and maintain some unreduced species in order to provide strong metal–support interaction to prevent sintering of active particles, on the other side.
 |
| | Fig. 2 H2-TPR profiles of the calcined catalysts. Dashed profiles correspond to bimetallic catalysts containing Fe and Ni, while solid lines indicate only Ni-based catalysts (the complex nature of materials and overlay of several reduction steps hamper a detailed interpretation). | |
Crystalline structure of calcined catalysts
All of the calcined catalysts showed patterns characteristic of the respective supports (Fig. 3). For catalyst 1, nickel oxide as well as hematite could be clearly identified. In addition, for catalyst 2, distinctive diffraction patterns could be assigned to either NiO crystals or MgO crystals. Both are face-centered-cubic and have very similar lattice constants (aNiO = 417 pm; aMgO = 421 pm). However, STEM/EDS measurements emphasize the formation of large NiO sponge-like crystals (Fig. 4). In all other cases, XRD analyses confirmed mainly X-ray amorphous materials potentially due to high dispersion of the active compounds on the amorphous supports.
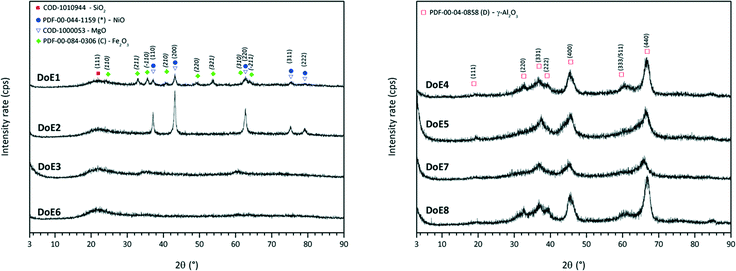 |
| | Fig. 3 X-ray powder diffractograms for the first calcined catalyst set. Diagrams are grouped for silica-based (left) and alumina-based catalysts (right). For the sake of clarity, the second set is not presented (very similar to the first set). | |
 |
| | Fig. 4 STEM/EDS area and point analyses of catalyst 2. Catalyst 2 Ni(15)/SiO2–MgO prepared by impregnation was analysed by STEM/EDS. The analysis shows the formation of large cubic sponge-like crystals. Area analyses (a) of the support and point measurements on the crystals (b) suggest that nickel is exclusively located in the sponge-like crystals. In contrast, magnesium is located in the support as well as in the NiO crystals. Error bars indicate the standard error of the mean. | |
Catalytic performance for methane production
Both sets were screened and compared with respect to carbon monoxide and propane conversion as well as methane and carbon dioxide selectivity after a time-on-stream of 1.5 h (Fig. 5). Catalyst 1 showed no activity due to pore blocking as confirmed by the physisorption measurements. Catalyst 2 was one of the most active catalysts but exhibited fast deactivation. This behavior can be assigned to sintering of the large porous crystals (Fig. 6) as well as pore infiltration by the formed coke deposits. Catalyst 8 displayed lower deactivation; sintering as well as coking could not be observed (Fig. 7). Furthermore, catalyst 7 showed lower activity than catalyst 5 most probably due to alloy formation (Fig. 8). The other four tested catalysts exhibited a very similar behaviour with overall low activity. The second set of catalysts, not presented in Fig. 5 for the sake of clarity, demonstrated comparable performance to the first set emphasising the reproducibility of the applied experimental methods.
 |
| | Fig. 5 Time course of carbon monoxide conversion for the first catalyst set. Catalytic tests were conducted at ambient pressure, a gas inlet temperature around 290 °C and a GHSV of 10400 h−1. The feed composition was set to CO:H2:C3H8 = 1.0:3.0:2.4. The feed as well as the output were monitored via on-line gas chromatography with 10 vol% N2 as an internal standard to account for volume contraction during the reaction. Catalysts 1 (♦), 3 (▲), 4 (×), and 6 (●) showed little activity. | |
 |
| | Fig. 6 HAADF STEM/EDS elemental mapping of catalyst 2 after the reaction. Severe sintering of the former sponge-like NiO crystals (Fig. 4) occurred. Color scale corresponds to intensity of corresponding X-ray emission line. | |
 |
| | Fig. 7 HAADF contrast in STEM of catalyst 8 before and after the reaction. The Ni(5)/Al2O3 catalyst shows an even metal dispersion (brightest dots) after calcination (a) which was maintained during the reaction (b). Furthermore, no carbonaceous deposits could be observed. | |
 |
| | Fig. 8 HAADF STEM/EDS elemental mapping of spent catalyst 7. The EDS analysis of catalyst 7 FeNi(15)/Al2O3–MgO prepared by precipitation provides evidence for the binary alloy formation of iron and nickel. Deactivation was caused by sintering to bigger particles. | |
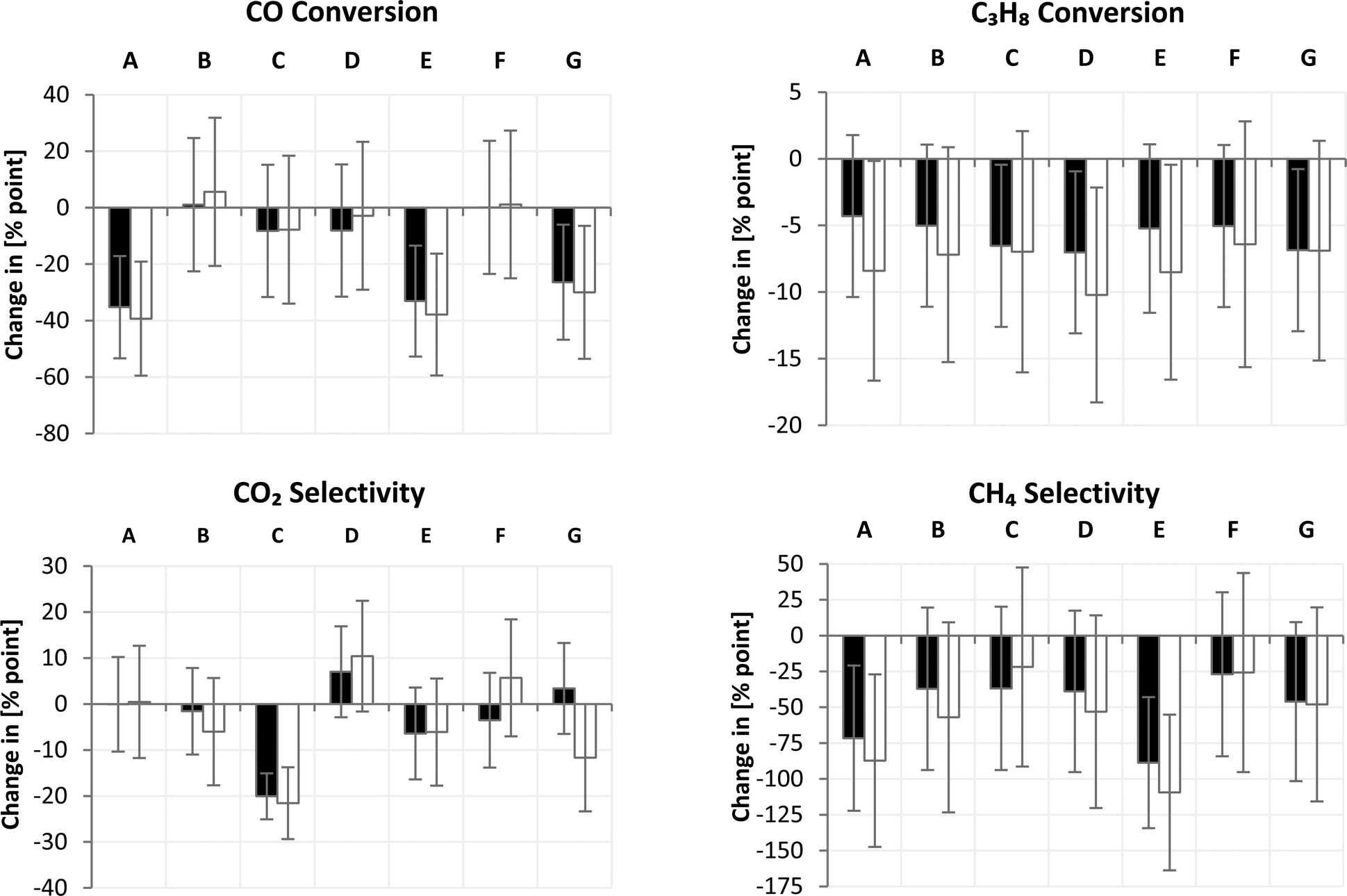
DoE analysis
For assessing the impact of the individual variables, A–G, and their respective levels, high and low, on each response, a full DoE analysis was performed as presented in Fig. 9. The impacts can be ranked according to their magnitudes. In addition, the variables can be differentiated according to their significant or negligible impact on the respective response (Table 5). Therefore, in the first step, the mean of each factor level consisting of 4 response values according to Table 2 has to be calculated including its standard error. In the second step, the difference between the upper and lower levels of each factor is calculated. The standard error of each impact is obtained by applying the propagation of uncertainty using the individual variances of the upper and lower factor levels (for formulas, see ESI† Section S5).
 |
| | Fig. 9 Impact of factors (A–G) on CO and C3H8 conversion as well as CH4 and CO2 selectivity for set 1 (■) and replica set 2 (□). Negative values refer to a decrease of the response, when a factor is changed from the lower level (−) to the upper level (+). The error bars refer to the standard errors calculated by error propagation of each factor level. Intersections with the base line correspond to impacts of no relevance. | |
Table 5 Ranking of important factors for controlling methanation with their preferred level in parenthesis
| Response |
Ranking |
| CO conversion |
A(−) > E(−) > G(−) |
| C3H8 conversion |
D(−) |
| CO2 selectivity |
C(−) |
| CH4 selectivity |
E(−) > A(−) |
By taking a look at CO conversion (Fig. 9), it becomes obvious that factor A caused the greatest change in conversion followed by factors E and G. All other factors exhibited a smaller influence or their influence was insignificant. The included bimetallic Fe–Ni-containing systems reached lower activity in CO conversion compared to sole nickel-based catalysts (factor A). This finding is contrary to expectations from the literature, where Fe:Ni = 1:1 is more active than monometallic nickel catalysts.28 Most probably, Fe–Ni alloy formation plays a crucial role but is difficult to address in a DOE approach. Alloy ratios of Fe:Ni = 1:1 and Fe:Ni = 3:1 are known to be active and have to be formed during reduction.29 Presumably, insufficient reduction to the metallic state could prohibit the right stoichiometric alloy formation since the Fe:Ni ratio is close to one (ESI† Section S4). Furthermore, potassium as a promoter (factor E) is known to lower activity by blocking active step sites even at very little content.30 In addition, the used amount of 5 wt% was quite high and pore blocking, as seen in physisorption measurements, disabled the methanation activity almost completely. Taking a look at different preparation methods (factor G), one can see that preparation via precipitation deteriorated CO hydrogenation. A reasonable explanation is the formation of a solid solution between MgO and NiO, depleting Ni from the Fe–Ni alloy as confirmed by STEM/EDS measurements (Fig. 8). The formation of a solid solution results in reduction at higher temperatures,29 as also observed in the TPR measurements (Fig. 2).
Propane conversion showed the same trend for both sets and for all variables; propane conversion was reduced when the factor was changed from the lower to the upper level. Factor D, in particular, showed the strongest impact. However, again, we have to be cautious in drawing quantitative conclusions bearing in mind the relatively high standard errors for all factors. As expected from catalytic hydrocracking,31 the use of a basic support resulted in lower hydrocarbon conversion, which is desirable for methanation in the presence of hydrocarbons.
Regarding CO2 selectivity, only factor C has a significant impact. The type of support or rather the basicity influences the CO2 adsorption capability.32 A less basic support such as SiO2 reduces CO2 formation.
The methane selectivity is mainly governed by the choice of active metal (factor A) and the amount of potassium as a dopant (factor E). Nickel is well known to possess high methanation activity at ambient pressure; whereas, iron has a more pronounced ability to form higher hydrocarbons.33,34 Therefore, the binary alloy of nickel and iron showed, besides the lower activity at ambient pressure, also lower methane selectivity. In addition, potassium is the preferred dopant for increasing the probability of chain growth to long-chained hydrocarbons at the expense of methane formation.33,34 On the other hand, potassium facilitates coke removal, improving catalyst stability.35 The high amount of potassium seemed to block most of the active sites resulting in low conversion and low methane selectivity. At these high levels, it was not possible to evaluate the positive effects of potassium on catalyst stability. Therefore, much lower dopant concentrations are required in order to establish the balance between the positive and negative influences of alkali promoters on catalyst performance in methanation.
The combination of the most significant factors and the preferred levels resulted in identifying a catalyst lead for further improvement. Therefore, we can conclude that, for CO methanation at ambient pressure and in the presence of propane as a LPG model compound, nickel-based systems with a low metal loading (5 wt%) on SiO2 prepared preferably by impregnation are the most appropriate catalysts. In addition, dilution with an inert material for better heat dissipation appears to be advantageous. The use of MgO or potassium compounds decreases the performance but should not be ruled out because these basic dopants are expected to enhance catalyst stability. Therefore, a lower dopant concentration will be considered in a follow-up design in order to establish the balance between positive and negative impacts on the overall catalyst performance.
Conclusions
Seven most studied factors in catalyst development concerning methanation were rated towards their impact on catalytic performance in downstream processing of residual syngas after LPG synthesis. The experimental effort could be reduced significantly by successfully applying a Plackett–Burman screening design. Most of the trends found in our study were in agreement with trends and expectations from conventional syngas chemistry and could be confirmed using several characterization techniques. The evaluation resulted in identification of the most relevant factors and pointed out the desirable levels. Pure nickel-based systems with low metal loading on SiO2 prepared by impregnation were identified as the most appropriate catalysts for further improvement. Therefore, this preliminary study opens the gates for the development of a superior catalyst lead for methanation of residual syngas in downstream processing after LPG synthesis.
Acknowledgements
This work was performed as part of a project with POSCO (South Korea) and the Research Institute of Industrial Science & Technology (RIST). The authors thank POSCO for financial support, Saint-Gobain NorPro for providing the silica support material (SS61137) as well as Sasol Germany GmbH for providing the alumina spheres (1.0/160).
Notes and references
- POSCO, POSCO Green Gas Tech established to generate clean energy, Press release, Korea, 07.04. 2014 Search PubMed.
- P. Sabatier and J. B. Senderens, C. R. Hebd. Seances Acad. Sci., 1902, 134, 689–691 Search PubMed.
- J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Fuel, 2010, 89, 1763–1783 CrossRef CAS.
- S. Takenaka, T. Shimizu and K. Otsuka, Int. J. Hydrogen Energy, 2004, 29, 1065–1073 CrossRef CAS.
-
J. R. Jennings, Catalytic Ammonia Synthesis: Fundamentals and Practice, Springer US, 2013 Search PubMed.
- H. C. Andersen and W. J. Green, Ind. Eng. Chem., 1961, 53, 645–646 CrossRef CAS.
- E. D. Park, D. Lee and H. C. Lee, Catal. Today, 2009, 139, 280–290 CrossRef CAS.
- A. Varone and M. Ferrari, Renewable Sustainable Energy Rev., 2015, 45, 207–218 CrossRef.
- S. Schiebahn, T. Grube, M. Robinius, V. Tietze, B. Kumar and D. Stolten, Int. J. Hydrogen Energy, 2015, 40, 4285–4294 CrossRef CAS.
- J. J. Gao, Y. L. Wang, Y. Ping, D. C. Hu, G. W. Xu, F. N. Gu and F. B. Su, RSC Adv., 2012, 2, 2358–2368 RSC.
- M. Agnelli, M. Kolb and C. Mirodatos, J. Catal., 1994, 148, 9–21 CrossRef CAS.
-
D. G. E. Kerfoot, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, DOI:10.1002/14356007.a17_157.
- R. L. Plackett and J. P. Burman, Biometrika, 1946, 33, 305–325 CrossRef.
- J. J. Gao, Q. Liu, F. N. Gu, B. Liu, Z. Y. Zhong and F. B. Su, RSC Adv., 2015, 5, 22759–22776 RSC.
- G. A. Mills and F. W. Steffgen, Catal. Rev.: Sci. Eng., 1974, 8, 159–210 Search PubMed.
- W. Wang, S. P. Wang, X. B. Ma and J. L. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- M. A. Vannice, Catal. Rev.: Sci. Eng., 1976, 14, 153–191 CAS.
- G. A. Somorjai, Catal. Rev.: Sci. Eng., 1981, 23, 189–202 CAS.
-
F. Fischer and H. Tropsch, Germany Pat., DE484337, 1927 Search PubMed.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and A. Ahmad, Green Chem., 2015, 17, 2647–2663 RSC.
- S. Tada and R. Kikuchi, Catal. Sci. Technol., 2015, 5, 3061–3070 CAS.
- J. Sehested, K. E. Larsen, A. L. Kustov, A. M. Frey, T. Johannessen, T. Bligaard, M. P. Andersson, J. K. Norskov and C. H. Christensen, Top. Catal., 2007, 45, 9–13 CrossRef CAS.
- B. C. Enger and A. Holmen, Catal. Rev.: Sci. Eng., 2012, 54, 437–488 CAS.
- D. C. Hu, J. J. Gao, Y. Ping, L. H. Jia, P. Gunawan, Z. Y. Zhong, G. W. Xu, F. N. Gu and F. B. Su, Ind. Eng. Chem. Res., 2012, 51, 4875–4886 CrossRef CAS.
- C. Mirodatos, H. Praliaud and M. Primet, J. Catal., 1987, 107, 275–287 CrossRef CAS.
-
E. Marceau, X. Carrier, M. Che, O. Clause and C. Marcilly, in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, DOI:10.1002/9783527610044.hetcat0022.
-
J. W. Geus and A. J. van Dillen, in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, DOI:10.1002/9783527610044.hetcat0021.
- M. P. Andersson, T. Bligaard, A. Kustov, K. E. Larsen, J. Greeley, T. Johannessen, C. H. Christensen and J. K. Norskov, J. Catal., 2006, 239, 501–506 CrossRef CAS.
- D. Y. Tian, Z. H. Liu, D. D. Li, H. L. Shi, W. X. Pan and Y. Cheng, Fuel, 2013, 104, 224–229 CrossRef CAS.
- H. S. Bengaard, J. K. Norskov, J. Sehested, B. S. Clausen, L. P. Nielsen, A. M. Molenbroek and J. R. Rostrup-Nielsen, J. Catal., 2002, 209, 365–384 CrossRef CAS.
-
D. S. Santilli and B. C. Gates, in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, DOI:10.1002/9783527610044.hetcat0088.
- C. K. Vance and C. H. Bartholomew, Appl. Catal., 1983, 7, 169–177 CrossRef CAS.
-
M. E. Dry, in Catalysis Science and Technology, ed. J. R. Anderson and M. Boudart, Springer-Verlag, Berlin, 1981, vol. 1, p. 159 Search PubMed.
-
M. E. Dry, in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, DOI:10.1002/9783527610044.hetcat0150.
-
B. E. Koel and J. Kim, in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, DOI:10.1002/9783527610044.hetcat0087.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description of analytical methods and the reactor setup. See DOI: 10.1039/c6re00071a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.