
A simple, one-pot synthesis of molybdenum oxide-reduced graphene oxide composites in supercritical methanol and their electrochemical performance†
Abstract
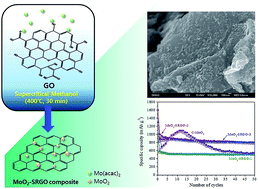
A simple and green supercritical methanol (scMeOH) route is developed to tightly anchor molybdenum oxide (MoO2) nanoparticles on reduced graphene oxide (RGO). In scMeOH, graphene oxide is reduced, and MoO2 nanoparticles with sizes of 10–20 nm are simultaneously deposited on the basal plane of RGO in a short time without using any reducing agents or additives. When tested as an anode in lithium ion batteries, the MoO2–RGO composites show enhanced electrochemical performance compared to bare MoO2. The composite with a MoO2 loading of 37.0 wt% delivers a high reversible discharge capacity of 793 mA h g−1 at 50 mA g−1 and an excellent rate performance of 205 mA h g−1 at 2.5 A g−1. After 100 cycles of high rate testing of up to 50 A g−1, the MoO2–RGO composite recovers most of its initial capacity. The improved electrochemical performance of MoO2–RGO can be attributed to the tight anchoring of nanosized MoO2 on RGO and the mesoporous structure of the composite. Consequently, the transport length of Li diffusion into the MoO2 phase is shortened, charge transfer kinetics at the electrode–electrolyte interface is facilitated, and the volume expansion associated with the conversion reaction can be accommodated.
 Please wait while we load your content...
Please wait while we load your content...

