
Synthesis and characterization of dimethyl 6-bromo-2H-chromene-2,3-dicarboxylate: thermodynamic and kinetics investigations by computational and experimental studies
Abstract
Modern society is dependent on synthetic chromene derivatives for use as drugs, including anticancer drugs, and for their biological activities. For this reason, the kinetics and synthesis of these compounds have attracted considerable attention, facilitating further developments and approaches for their synthesis. In this study, dimethyl 6-bromo-2H-chromene-2,3-dicarboxylate (4) was synthesized via the reaction between triphenylphosphine (1), dimethyl acetylenedicarboxylate (2) and 5-bromo-2-hydroxybenzaldehyde acid (3) in the presence of dichloromethane, and it was then characterized using IR, 1H, 13C, and 31P NMR. The kinetics and mechanism of the reaction were theoretically and experimentally investigated using the stopped-flow and UV-vis spectrophotometry approaches. The reaction mechanism involved a number of steps, starting with the fast reaction between reactants 1 and 2 to generate I1, and this step was investigated using stopped-flow apparatus. The consumption of the intermediate 1 (I1) and 3 was studied using a UV-vis technique and it was found to follow a first-order kinetics. The partial order of compound 3 was determined to be zero and had no effect on the rate of the reaction. The kinetics data showed that step4 of the proposed mechanism was the rate-determining step. Investigations of the consumption of I1 at different temperatures allowed the activation parameters to be specified with respect to the slowest step of the proposed mechanism using two linearized forms of the Eyring equation. From the temperature, concentration, and solvent studies, the activation energy (Ea = 61.30 kJ mol−1) and the related activation parameters (ΔG‡ = 78.42 ± 4.61 kJ mol−1, ΔS‡ = −67.09 ± 7.96 J mol−1 and ΔH‡ = 58.88 ± 2.34 kJ mol−1) were calculated. Theoretical investigations were performed for further understanding the proposed mechanism at the B3LYP/6-31++g(d,p) and M06/6-31++g(d,p) levels. The theoretical and experimental data indicated that the rate of the overall reaction was second-order, and depended on the concentrations of compounds 1 and 2. The proposed mechanism was confirmed with the observed kinetics data obtained from the computational and experimental studies.
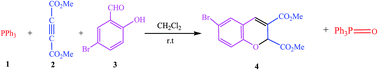
 Please wait while we load your content...
Please wait while we load your content...

