
Selective removal of IgG from the urine of patients with proteinuria using a polymer coated core–shell magnetic nanoparticle†
Zhifen Denga,
Kai Hub,
Liangliang Bib,
Hang Yuan*c,
Yanlong Chena,
Shengnan Zhaoa,
Huifang Dua,
Xuesheng Yuana,
Yanjie Huang*b and
Shusheng Zhang*ac
aCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450001, P. R. China. E-mail: zsszz@126.com
bHenan University of Chinese Medicine, Zhengzhou 450008, P. R. China. E-mail: huangyanjie69@hotmail.com
cCenter for Advanced Analysis and Computational Science, Zhengzhou University, Zhengzhou, 450001, P. R. China
First published on 26th October 2016
Abstract
Selective removal of highly abundant proteins prior to analysis is an important issue in proteomics. In this study, a core–shell structured magnetic nanocomposite of Fe3O4@SiO2@MAsp-HEMA was found to be an effective and selective adsorbent to remove highly abundant IgG from the urine of patients with proteinuria. This novel magnetic adsorbent was prepared by coating a poly(N-methacryloyl-L-aspartic acid-hydroxyethyl methacrylate) shell on Fe3O4@SiO2 nanoparticles via a coupling agent of KH-570 using the mini-emulsion polymerization method. The as-synthesized nanoparticle was characterized by X-ray diffraction, elemental analysis, thermal gravimetric analysis, infrared spectroscopy, magnetic hysteresis loop curves, scanning electronic microscopy and transmission electron microscopy. A kinetic adsorption experiment was performed to investigate the speed of adsorption and the time to reach the adsorption balance. 1D SDS-PAGE analysis and capillary electrophoresis were used to validate its selective adsorption ability. The resulting Fe3O4@SiO2@MAsp-HEMA showed a uniform spherical shape at nanoscale dimensions of about 200–300 nm, removing efficiency above 80%, and maximum adsorption capacity of 18.08 μg mg−1 at 15 ± 0.5 °C. The binding between IgG and the ligand was confirmed by isothermal titration calorimetry. The hydrogen bonding force was demonstrated as one of the major interactions between the functional groups of the as-synthesized magnetic nanoparticles and IgG by a molecular docking experiment.
Introduction
Immunoglobulin G (IgG) is a pivotal glycoprotein and functions as an antibody in immune system responses. Structurally, IgG is a large Y-shaped molecule with a molecular weight of approximately 150 kDa and contains four polypeptide chains (two heavy chains (50 kDa) and two light chains (25 kDa)), which are connected by disulfide bonds. IgG has always been purified and enriched for diagnoses and therapies in clinical studies. However, a trouble role of IgG is as a kind of high-abundant protein of biofluids (e.g. urine) in proteomics.1Bodily fluids are regarded as the best sources of protein for the identification of biomarkers of disease in proteomics, whereas the main drawback of the comprehensive analysis of bodily fluids is the high abundance of albumin and IgG. The presence of these proteins can obscure other low abundance proteins in one-dimensional electrophoresis (1DGE), two-dimensional gel electrophoresis (2DGE), and liquid chromatography (LC) separation and suppress the signals of low abundance proteins in LC-MS/MS identification.2 Depletion of these highly abundant proteins is a way to enhance the sensitivity and resolution of analyses.3 Commercial albumin/IgG removal kits have been widely used for plasma and cerebrospinal fluid samples based on the recognition of protein A or G affinity ligands;4 however, these methods have not been extensively studied in large volume samples such as urine. In a recent study, highly abundant proteins were depleted in urine samples by commercially available immunodepletion and ion-exchange based approaches for the enrichment of low abundance proteins. The results showed that no clear advantage of protein depletion was observed in low abundance protein identification.1 Moreover, using such depletion kits is not economical or operationally friendly. Taking these factors into account, developing a new affinity ligand for the removal of these highly abundant proteins from bodily fluids, which is convenient, economical and efficient with less negative effects, is challenging.
Recombinant antibodies are well recognized affinity agents. However, their expensive cost, long-time consumption and imperfect applicability limited the development and application of such an approach. Some alternates including peptides,5 aptamers,6 and chemical ligands7 expand the candidate pool of protein capture reagents. We have been developing an alternative approach for protein IgG capture based on the observation that the active sites on the target protein are complementary to the ligands and the link are some weak interactions such as hydrogen bonding, electrostatic interactions and van der Waals.8
Recently, a functional monomer N-methacryloyl-L-aspartic acid (MAsp) as a synthetic ligand showed affinity to IgG,9 which attracted our interest to utilize its specificity to prepare a functional polymer composite Fe3O4 magnetic nanoparticle to remove IgG from urine.
Polymer composite Fe3O4 nanoparticles are widely studied magnetic nanoparticles (MNPs). Due to their super paramagnetism and functional groups on their surface, Fe3O4 MNPs have been a good carrier to selectively capture targets and then be quickly separated from a complex matrix with a magnet. Nowadays, Fe3O4 MNPs have been widely applied for small and large molecular enrichment and separation from environmental, food, biological samples.10–12
Although urine analysis is a noninvasive method for kidney disease biomarker screening, highly abundant proteins are a major interference. In this study, our aim was to prepare poly(MAsp-HEMA) composite Fe3O4 nanoparticles via a magnetic separation method to remove IgG from large volumes of urine. The interaction and binding affinity between IgG and MAsp was studied by a molecular docking study, isothermal titration calorimetry (ITC), and adsorption kinetics. The adsorption selectivity was demonstrated by one-dimensional dodecylsulfate polyacrylamide gel electrophoresis (1D SDS-PAGE) and capillary electrophoresis (CE).
Experimental
Chemicals and reagents
Analytical grade ferric chloride hexahydrate (FeCl3·6H2O), ethylene glycol (EG), anhydrous sodium acetate (NaAc), polyethylene glycol (PEG, Mw 8500–11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), ammonium hydroxide (NH3·H2O, 28 wt%) and tetraethyl orthosilicate (TEOS, >99%, GC purity, Aladdin, Shanghai, China) were used to prepare Fe3O4@SiO2 nanoparticles. Hydroxyethyl methacrylate (HEMA) and ethylene glycol dimethacrylate (EGDMA) were purchased from Aladdin biological technology Co., Ltd. (Shanghai, China). Di-tert-butyl-L-aspartate hydrochloride (DTAH), methacryloyl chloride, and 3-(trimethoxysilyl)propyl methacrylate (KH-570, stabilized with BHT) were purchased from Tianjin Heowns Biochemical Technology Co., Ltd. (Tianjin, China). Immunoglobulin G (IgG, 5 mg mL−1, 0.01 M PBS, pH 7.2) was obtained from Antu biotechnology Co., Ltd. (Zhengzhou, China). Bovine serum albumin (BSA) powder was purchased from Solarbio (Beijing). Water used was purified by a Milli-Q purification system. N,N,N′,N′-Tetramethylene diamine (TEMED), acrylamide (30%), sodium dodecyl sulfate (SDS, 10%, w/w), ammonium persulfate (APS, 10%, w/v), 1.5 M tris–HCl buffer (pH 6.8) and 1.0 M tris–HCl buffer (pH 8.8) were used to prepare the polyacrylamide gel. A Thermo 26616 page ruler prestained protein ladder (10–170 kDa) was used as a marker.
000), ammonium hydroxide (NH3·H2O, 28 wt%) and tetraethyl orthosilicate (TEOS, >99%, GC purity, Aladdin, Shanghai, China) were used to prepare Fe3O4@SiO2 nanoparticles. Hydroxyethyl methacrylate (HEMA) and ethylene glycol dimethacrylate (EGDMA) were purchased from Aladdin biological technology Co., Ltd. (Shanghai, China). Di-tert-butyl-L-aspartate hydrochloride (DTAH), methacryloyl chloride, and 3-(trimethoxysilyl)propyl methacrylate (KH-570, stabilized with BHT) were purchased from Tianjin Heowns Biochemical Technology Co., Ltd. (Tianjin, China). Immunoglobulin G (IgG, 5 mg mL−1, 0.01 M PBS, pH 7.2) was obtained from Antu biotechnology Co., Ltd. (Zhengzhou, China). Bovine serum albumin (BSA) powder was purchased from Solarbio (Beijing). Water used was purified by a Milli-Q purification system. N,N,N′,N′-Tetramethylene diamine (TEMED), acrylamide (30%), sodium dodecyl sulfate (SDS, 10%, w/w), ammonium persulfate (APS, 10%, w/v), 1.5 M tris–HCl buffer (pH 6.8) and 1.0 M tris–HCl buffer (pH 8.8) were used to prepare the polyacrylamide gel. A Thermo 26616 page ruler prestained protein ladder (10–170 kDa) was used as a marker.
Apparatus
A Teflon-lined stainless-steel autoclave (PSK-100 mL, Binhai Zhengxin Instrument Factory) and a mechanical agitator were used for the preparation of magnetic nanoparticles. High purity nitrogen (≥99.999%) was used to protect the reaction. An ultrasonic cell disruptor and KQ 5200 ultrasonic cleaner (Kunshan Ultrasonic Instrument Co., China) were used for mini-emulsion polymerization. A Bio-Rad vertical slab gel electrophoresis apparatus was used for 1D SDS-PAGE. Capillary electrophoresis experiments were performed using the HP3DCE system equipped with a diode-array detector and Chemstation software (Agilent Technologies, Waldbronn, Germany). An ITC200 microcalorimeter from MicroCal, LLC (Northampton, MA) was used for the ITC experiment.Synthesis of N-methacryloyl-L-aspartic acid (MAsp) monomer
MAsp was prepared by two simple steps. First, in a 25 mL round-bottom flask, DTAH (1 mmol) was dissolved in 8 mL of anhydrous dichloromethane (DCM) and stirred magnetically, and then anhydrous triethylamine (TEA, 450 μL) and methacryloyl chloride (150 μL) were successively added into this solution with stirring for 24 h at room temperature. The final solution was rotary evaporated under reduced pressure and then the crude product was purified by column chromatography (eluent: petroleum ether/ethyl acetate (6:1, v/v)) and rotary evaporated to give product A, which is obtained as a white crystalline solid. +ESI/MS (m/z): [M + H]+ 314.3, [M + Na]+ 336.4, [M + K]+ 352.4; 1H NMR (400 M Hz, CDCl3): δ (ppm) = 1.46 (s, 9H, –C(CH3)3), 1.47 (s, 9H, –C(CH3)3), 1.98 (s, 3H, –CH3), 2.74–2.79 (dd, J1 = 17 Hz, J2 = 4.36 Hz, 1H, –CH2–), 2.89–2.94 (dd, J1 = 17 Hz, J2 = 4.14 Hz, 1H, –CH2–), 4.71–4.73 (m, 1H, –CHCH2–), 5.37 (s, 1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.77 (s, 1H, CH2), 6.82, 6.84 (d, J = 8.0 Hz, 1H, –NH–) (Fig. S1†).
CH2), 5.77 (s, 1H, CH2), 6.82, 6.84 (d, J = 8.0 Hz, 1H, –NH–) (Fig. S1†).
In the second step, 1 mmol of product A was dissolved in 1.2 mL of DCM and 1.2 mL of trifluoroacetic acid (TFA) was subsequently added with magnetic stirring for 9 h at room temperature with a TLC monitor. Then, the solution was rotary evaporated with DCM for some time and an oil pump was used to extract the residual organic solvent for 1 h. Then, the yellow solid MAsp was obtained. +ESI/MS (m/z): [M + H]+ 202.3, [M + Na]+ 224.3, [M + K]+ 240.3; 1H NMR (400 M Hz, DMSO), the active hydrogen of this carboxylic acid compound cannot occur in this spectrum. δ (ppm) = 1.848 (s, 3H, –CH3), 2.58–2.79 (dd, J1 = 61.29 Hz, J2 = 16.18 Hz, 2H, –CH2–), 4.56–4.62 (m, 1H, –NHCHCH2–), 5.38 (t, 1H, CH2), 5.69 (t, 1H, CH2), 8.14, 8.16 (d, J = 8.19 Hz, 1H, –NH–), 5.7530 (DCM residue in DMSO) (Fig. S2†). MAsp was stored in a refrigerator at 4 °C.
Preparation of Fe3O4@SiO2@MAsp-HEMA
First, the Fe3O4 nanoparticles were prepared by means of solvothermal synthesis, according to the method reported,10,13 and then the obtained Fe3O4 from the first step was used for the preparation of Fe3O4@SiO2 by a sol–gel method. To introduce a double bond for polymerization, KH-570 was chosen as the coupling agent to prepare Fe3O4@SiO2@KH-570 in the third step. Last, a mini-emulsion polymerization method14 was adopted to prepare Fe3O4@SiO2@MAsp-HEMA. HEMA was the monomer, EGDMA was the cross-linker, and SDS and PVA consisted of a complex emulsifier. (NH4)2S2O8 and NaHSO3 were used as the redox initiators. The detailed procedure is presented in the ESI.†Characterizations
The morphology of the magnetic nanoparticles was characterized by Scanning Electron Microscopy (SEM, SEM-6510, Japan) and Transmission Electron Microscopy (TEM, FEI Tecnai G2 F20, The Czech Republic). The crystallinity and phase purity of the products were examined using X-ray diffraction (XRD) in an X-ray diffractometer (XRD, X'Pert PRO, PANalytical, The Netherlands) with Cu Kα (40 kV, 40 mA) radiation (k = 1.5406 Å). Fourier transform infrared spectra (FTIR) (Bruker Vector 22 instrument) were obtained to confirm the presence of functional groups on the magnetic particles. The bonded amount was determined by Elementary Analysis (EA, EA 1112 elemental analyser) and thermogravimetric analysis (TGA, Shimadzu DT-40 thermal analyser, test range: 20–800 °C, heating rate: 10 °C min−1, argon atmosphere). The thermal stability of magnetic nanoparticles was also measured by TGA. The magnetic measurements were performed at 300 K using a MPMS-XL-7 magnetometer (Quantum Design Inc., USA) to test the saturation magnetization.Molecular-docking study
The structure of MAsp was constructed using a ChemOffice 2004 software. The structure was energy minimized using the steepest descent and conjugate gradient techniques. The 3D crystal structure of the human IgG Fc fragment (1FC2) was downloaded from the RCSB PDB Protein Data Bank. Docking was performed using the automated docking tool of an AutoDock software 2.5 according to the genetic algorithm method with some modifications. The docking runs were carried out in an active pocket where the original ligand was located.Isothermal titration calorimetry (ITC)
The experiments were performed in a PBS (0.01 M, pH 7.2) buffer. The concentration of IgG and BSA was 55 μM and MAsp was 1 mM. In each individual experiment, about 38 μL of the MAsp solution was injected through the computer controlled 40 μL microsyringe at an interval of 150 s into the protein solution (cell volume = 200 μL) with stirring at 750 rpm at 25 °C. The experimental data were fitted into a theoretical titration curve using the software supplied by MicroCal. A standard one-site model was used with ΔH (enthalpy change, kcal mol−1), Ka (association constant, M−1), and N (number of binding sites) as the variables.Kinetic adsorption test
In the kinetic adsorption test, 10 mg of Fe3O4@SiO2@MAsp-HEMA MNPs was suspended in 5 mL PBS (0.01 M, pH 7.2) with IgG at a concentration of 0.04 mg mL−1 and shaken on a shaking table at 120 rpm at a constant temperature (15 ± 0.5 °C) for 5 min to 48 h. Then, the supernatants and MNPs were separated for 3 min by an external magnetic field and the concentration of IgG in the filtrate was determined using an ultraviolet spectrophotometer at 280 nm. The adsorption amounts (Q, mg g−1) of Fe3O4@SiO2@MAsp-HEMA were calculated according to eqn (1), and the pseudo-second-order rate kinetic model was applied to fit the kinetic data according to eqn (2),
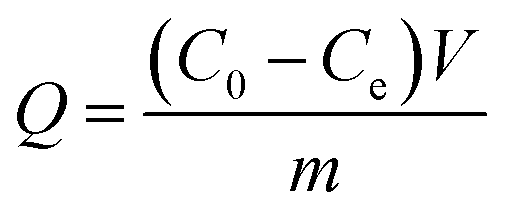 | (1) |
 | (2) |
Selectivity assay
Three samples (containing proteinuria) of pediatric Henoch Schonlein purpura nephritis (HSPN, Sample 1, Sample 2 and Sample 3) were obtained from the Pediatrics Department of the First Affiliated Hospital of Henan University of Chinese Medicine. Two real samples were directly used to test the selectivity of the polymer coated MNPs by 1D SDS-PAGE.Details areas follows: proteinuria (15 mL) was centrifuged at 5000 g for 20 min to separate the suspended matter. Then, 30 mg of preconditioned Fe3O4@SiO2@MAsp-HEMA MNPs with PBS was added, followed by vortex stirring for 1 min, contact for 2 h, and then a magnet was used to remove the MNPs. The MNPs treated urine was then filtered by a NC membrane15 and the proteins were adsorbed on the NC membrane. The NC membrane was cut into small pieces, and then acetone was simultaneously used to dissolve the membrane and precipitate the protein under ultrasonication. The sample was centrifuged at 14000g for 30 min to achieve the protein precipitation. The sample was dried under air of a super clean bench and 8 M lysis buffer was used to redissolve the sediment. The protein concentration was determined by the Coomassie brilliant blue method. Another same sample was treated according to the process abovementioned without the MNPs treatment. Then, 25 μg of protein was used to perform the reduced 1D SDS-PAGE.
Moreover, field-amplified sample stacking capillary electrochromatography (FASS-CE) was also used to verify the removal efficiency of the as-synthesised MNPs. Sample 3 and its IgG spiked sample and an IgG spiked sample treated with MNPs were analysed. The optimal electrophoretic condition was as follows: background electrolyte, 2.3 M acetic acid + 0.05% Tween20 (pH = 2.1); a bare fused silica capillary (Yongnian Optical Fiber Corporation, Hebei Province, China, 55 cm × 50 μm i.d.); separation voltage, 10 kV; column temperature, 25 °C; UV detection wavelength, 280 nm; water-plug injection: 30 mbar × 30 s; and sample electrokinetic injection, 25 kV × 30 s. The capillary was flushed with the buffer solution for 3 min before each injection and flushed with 100 mM NaOH and water for another 3 min after each separation run.
Results and discussion
Characterization results
According to the FTIR spectrum (Fig. 1), unique peaks of –Si–O–, –CH3–, –CH2–, –CO– were obtained for Fe3O4@SiO2@KH-570 as compared with those for Fe3O4 and Fe3O4@SiO2. For Fe3O4@SiO2@MAsp-HEMA, the absorption peaks of –CO–, –CH3–, and –CH2– at 1726.23, 2890.24 and 2956.95 cm−1 obviously increased, which indicates that polymerization had successfully occurred out of the core of the Fe3O4 magnetic nanoparticles.
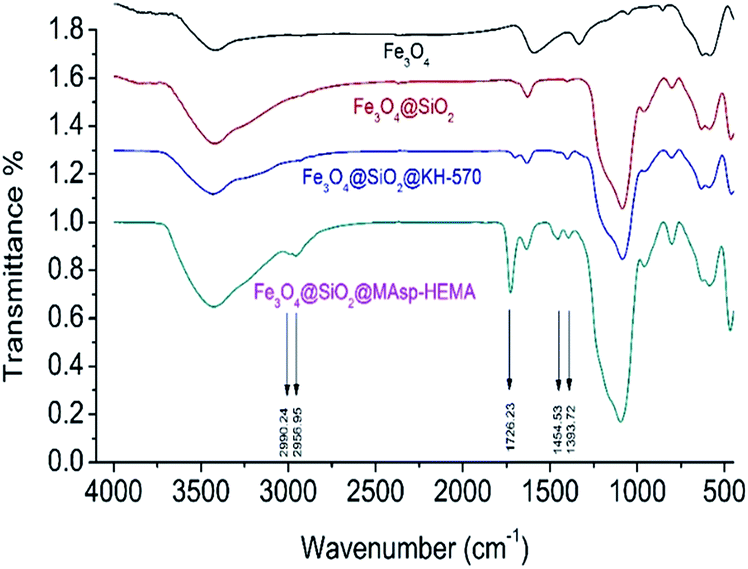 | ||
| Fig. 1 The IR spectrum of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@KH-570 and Fe3O4@SiO2@MAsp-HEMA. | ||
According to the EA results (Table 1), it can be seen that KH-570 was successfully bonded to Fe3O4@SiO2, and the bonded amount is 353.3 μmol g−1 calculated by the carbon content. In addition, the content of carbon and hydrogen of Fe3O4@SiO2@MAsp-HEMA is higher than in Fe3O4@SiO2@KH-570, which indicates that the polymerization was successful.
| Samples | C% | H% |
|---|---|---|
| Fe3O4@SiO2 | 0 | 0 |
| Fe3O4@SiO2@KH-570 | 4.24 | 1.04 |
| Fe3O4@SiO2@MAsp-HEMA | 12.96 | 2.12 |
The thermo gravimetric curve of Fig. S3† shows that the decomposition temperature of Fe3O4@SiO2@MAsp-HEMA is at 251.9 °C, and the mass loss occurred in the range of 251.9–800 °C, which demonstrates the good thermal stability of Fe3O4@SiO2@MAsp-HEMA.
The crystalline structure and phase purity were characterized by XRD. As shown in Fig. S4,† all the diffraction peaks of Fe3O4@SiO2, Fe3O4@SiO2@KH-570 and Fe3O4@SiO2@MAsp-HEMA can be indexed to the magnetic cubic structure of the Fe3O4 phase (JCPDS 75-1609) without any other impurity peaks. At 2θ = 22.910, the wide dispersion peak corresponds to amorphous SiO2. Moreover, the diffraction peaks show relative broadness and low intensity, indicating that the crystalline size of these particles is on the nanoscale.
According to the magnetic hysteresis loop curves in Fig. 2A, all the MNPs show superparamagnetic behavior without obvious remanence or coercivity at 300 K. Saturation magnetization values of 79.8 emu g−1 (Fe3O4), 76.0 emu g−1 (Fe3O4@SiO2), and 34.1 emu g−1 (Fe3O4@SiO2@MAsp-HEMA) were determined. The polymer layer coated on the shell of Fe3O4 resulted in a drop in the saturation magnetization due to an increase in the total mass of the organic shell.
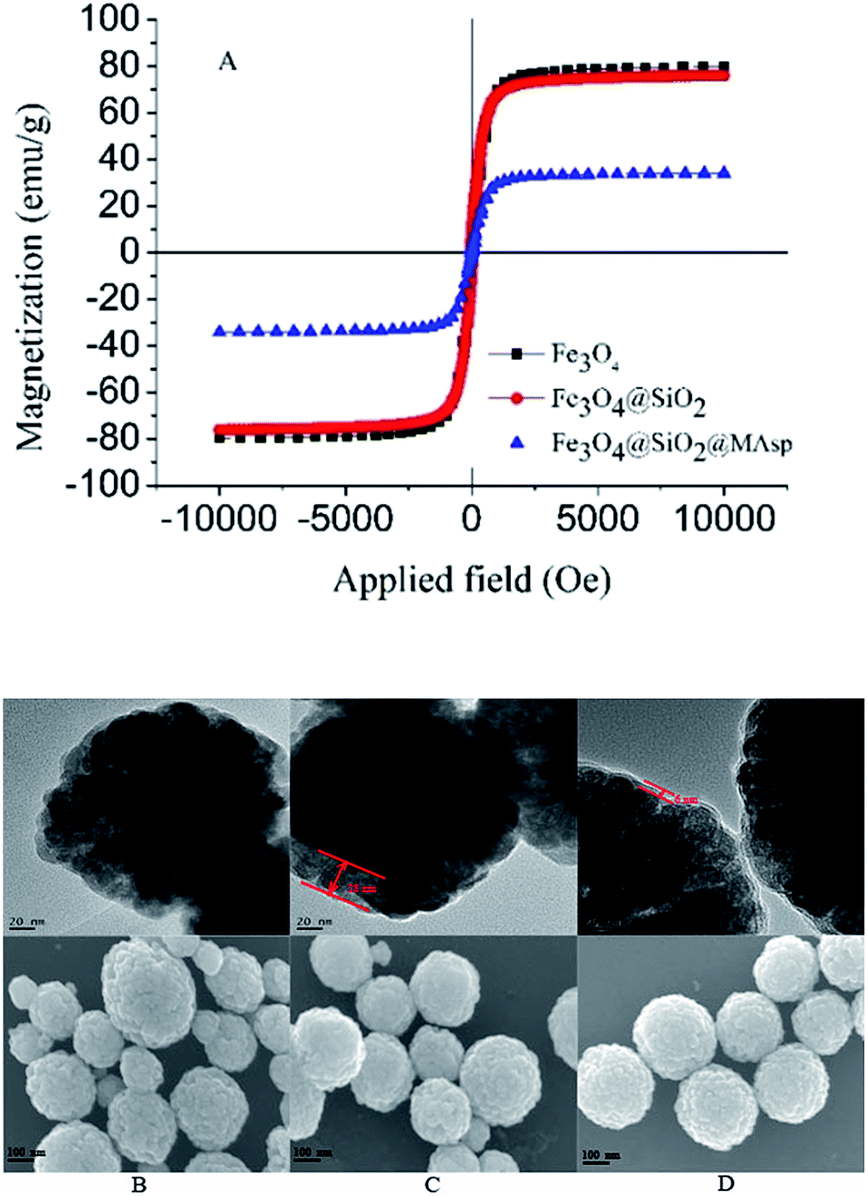 | ||
| Fig. 2 Magnetic hysteresis loops of Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2@MAsp-HEMA (A), the TEM (upper) and SEM (lower) images of Fe3O4 (B), Fe3O4@SiO2 (C) and Fe3O4@SiO2@MAsp-HEMA (D). | ||
The modification of the Fe3O4 nanoparticles with a SiO2 shell is essential for the coating of the subsequent MAsp-HEMA layer. According to Fig. 2B–D, it is obvious that the core–shell structure was obtained, and the nanoparticles are globular and uniformly dispersed without aggregation. The size of the as-synthesized MNPs is about 307 ± 61 nm and the SiO2 shell is almost 32 ± 4 nm. Interestingly, all the MNPs show peculiar cauliflower-like morphology, which is different from the reported images of the Fe3O4 composite nanoparticles. Nanomaterials with a cauliflower-like structure have been reported recently, which generally have a high specific surface area, photocatalytic activity, magnetism, light absorption, and microwave absorbing ability.16–22 We inferred that this morphology is related to the heating rate and the polymerization degree of PEG, as these parameters were different from the reported ones.
Molecular-docking study
We performed a molecular-docking study to confirm and gain more insight into the interaction of MAsp with the IgG Fc fragment using an automatic docking tool of Auto Dock 2.5 software. The structure of the IgG Fc fragment was obtained from the Protein Data Bank (PDB).According to the computing screen, the IgG Fc fragments contained four active site pockets that can bind to MAsp. The best site is shown in Fig. 3a with a binding energy of −4.48 kcal mol−1, inhibition constant of 0.519 nM, and total internal energy of −8.13 kcal mol−1. Two hydrogen bonds are formed (Fig. 3b) and the bonding energies are −5.202 and −1.371 kcal mol−1, respectively. These values indicate that Phe241, Phe242 and Lys334 residues may play major roles in the IgG Fc binding.
 | ||
| Fig. 3 Docking results for the interaction of MAsp with the IgG-Fc fragment (PDB 1FC2). (a) 3D structure of Fc-MAsp complex at the optimal binding site (b) details of MAsp (stick model) interaction with Fc fragment. Dotted lines indicate hydrogen bond formation. | ||
Isothermal titration calorimetry
Molecular-docking was performed to study the theoretical binding affinity. The real binding affinity was measured by isothermal titration calorimetry (ITC). In the ITC experiment, the MAsp solution was loaded into the syringe and titrated with the protein solution in the calorimeter cell. Upon binding between the protein (IgG or BSA) and MAsp, heat is released. The amount of heat generated decreased with each subsequent injection as MAsp in the cell became saturated. In contrast, if no binding occurs, the released heat, simply caused by reagent mixing, is much smaller and essentially remains constant with each injection. Fig. S5(a)† is a typical titration experiment of MAsp with IgG at pH 7.2 in the PBS buffer. As expected, the heat generated at each injection of MAsp gradually decreased with each additional injection, yielding a typical titration isotherm. The enthalpy change (ΔH) is −928.9 ± 91.14 cal mol−1, the entropy change (ΔS) is 19.2 cal mol−1 deg−1, the number of IgG binding sites is 1.84 ± 0.118, and the association constant (Ka) is 7.39 ± 2.76 × 104 M−1. In a contrast experiment, Fig. S5(b)† shows that no binding occurs between BSA and MAsp. This result is similar to Fig. S5(c)† in which no protein was present in the PBS buffer. The heat release was small and almost unchanged during the titration. Therefore, the result of an ITC experiment is essentially in agreement with that of the molecular-docking and the binding between IgG and MAsp is selective compared to randomly selected BSA.Adsorption kinetics
The pseudo-second-order rate kinetic models were used to determine the rate-controlling and mass transfer mechanisms. The parameter (t/Qt) was plotted against t (min) and k2 and Qe were determined using the intercept and slope of the linear plot, respectively. The value of k2 is 0.746 mg μg−1 min−1, Qe is 18.08 μg mg−1, and R2 = 1, which showed that the pseudo-second-order model fit the experimental data well. As seen in Fig. 4, rapid adsorption takes place in the first 2 h. Subsequently, the adsorption becomes slow and almost reaches an equilibrium within 12 h. No appreciable removal takes place, indicating that complete adsorption occurs within 2 h, thus subsequent adsorption was conducted for 2 h. The maximum adsorption efficiency reached 90.1%. | ||
| Fig. 4 Adsorption kinetics of Fe3O4@SiO2@MAsp-HEMA. | ||
Selectivity assay
To test the biospecific nature of the polymer–IgG binding, we conducted an adsorption experiment by incubating Fe3O4@SiO2@MAsp-HEMA in a real urinary protein mixture and evaluated the extent of protein adsorption via 1D SDS-PAGE. The urine samples we chosen in this experiment are human proteinuria. This kind of sample is frequently used for biomarker discovery of kidney disease. Thus, these are good and direct samples to test the material's specificity to adsorb IgG. According to Fig. 5a of the standard control, there are two bands. One is the heavy chain of the reduced IgG (55 kDa) and another band is its light chain (25 kDa). In Fig. 5b, S1 and S2 stand for two different proteinuria samples, whereas tre-S1 and tre-S2 stand for samples treated by the as-synthesized MNPs. There are three main bands that exist in each lane of S1 and S2, whereas for tre-S1 and tre-S2, two bands (55 kDa and 25 kDa corresponding to IgG) obviously decreased compared to the MNPs in the untreated samples. The other bands almost did not decrease, which indicates that these as-synthesized MNPs can selectively remove IgG from the protein mixture. | ||
| Fig. 5 1D SDS-PAGE. (a) Standard IgG; (b) two samples (s1, s2) and corresponding MNP treated samples (tre-s1, tre-s2). Elliptic curves mark the changed bands. | ||
Since IgG is a large molecule, a conventional HPLC method was required to use the protein analysis column. In terms of ease and economy, the FASS-CE method was adopted to verify this specific adsorption. While proteins tend to adsorb in the silica tube, it is necessary to establish a method for the determination of IgG. The details of the method optimization experiments are in the ESI.† Under optimal conditions, sample 3 (HSPN), IgG spiked sample, and MNPs treated IgG spiked sample were analysed. The retention time of IgG was 2.01 min, as shown in Fig. 6. After treatment with MNPs, the peak of IgG became smaller and other peaks were not changed. The removal efficiency was near 80%, which indicates the specificity of Fe3O4@SiO2@MAsp-HEMA to IgG.
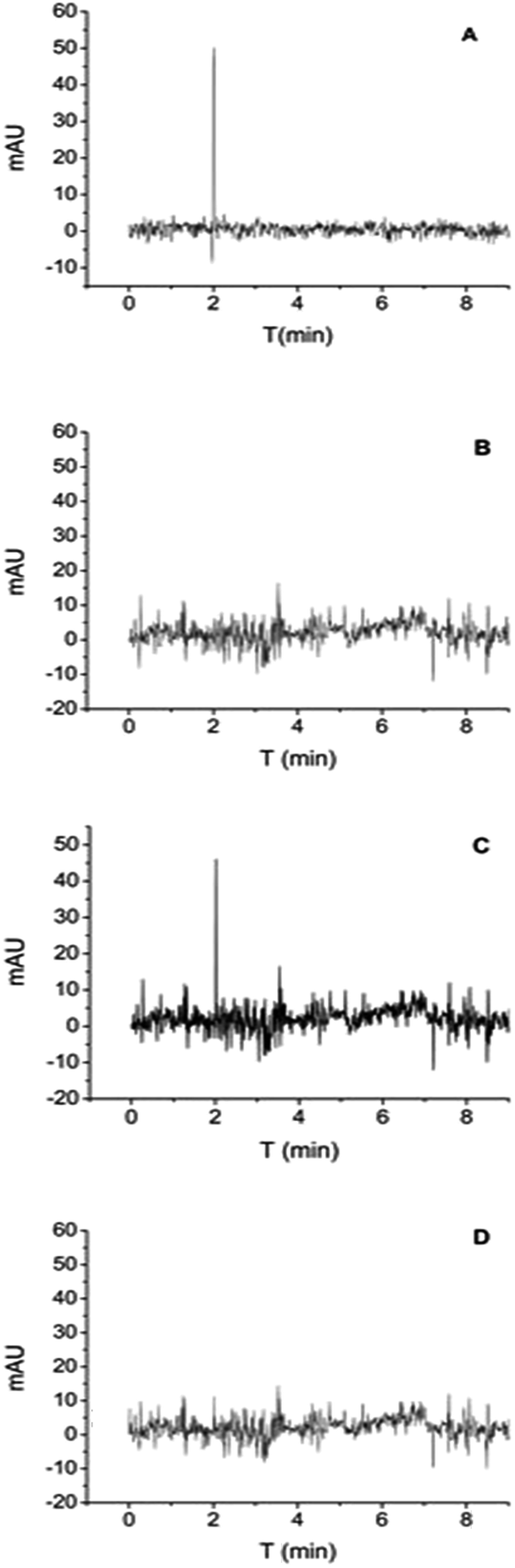 | ||
| Fig. 6 CE spectrum of IgG standard solution (A), sample 3 (B), IgG spiked sample 3 (C), MNPs treated IgG spiked sample 3 (D). | ||
Conclusions
In summary, a robust synthetic route for the preparation of Fe3O4@SiO2@MAsp-HEMA nanoparticles with a core–shell structure was presented. Characterization results showed that the morphology of the as-synthesized MNPs was cauliflower-like, which may provide a large specific surface area. A kinetic adsorption experiment showed that rapid adsorption occurred in the first 2 h. 1D SDS-PAGE and CE experiments confirmed that Fe3O4@SiO2@MAsp-HEMA nanoparticles have high specificity and selectivity. A molecular docking experiment revealed that the interaction between the functional group of MAsp and IgG Fc fragment was mainly hydrogen bonding. An ITC experiment confirmed the existence of binding between IgG and MAsp. It is expected that the as-synthesized Fe3O4@SiO2@MAsp-HEMA holds a great potential in urinary proteomic research, whereas the shortcoming is that the MAsp ligand number should be investigated to provide more interaction sites in the polymer shell to further enhance the removal efficiency.Acknowledgements
The authors acknowledge the support of the National Natural Science Foundation of China (21475119, 21275133 & 21605042).Notes and references
- S. Filip, K. Vougas, J. Zoidakis, A. Latosinska, W. Mullen, G. Spasovski, H. Mischak, A. Vlahou and J. Jankowski, PLoS One, 2015, 10, 20 Search PubMed.
- E. P. Diamandis, Mol. Cell. Proteomics, 2004, 3, 367–378 CAS.
- L. Zou, W. Li, D. Wang, Z. Wang and W. Sun, J. Basic Clin. Med., 2015, 35, 439–443 Search PubMed.
- H. Li, R. Ortiz, L. B. Tran, H. Salimi-Moosavi, J. Malella, C. James and J. Lee, AAPS J., 2013, 15, 337–346 CrossRef CAS PubMed.
- Y. X. Zhang, N. Islam, R. G. Carbonell and O. J. Rojas, ACS Appl. Mater. Interfaces, 2013, 5, 8030–8037 CAS.
- K. Kim, S. Lee, S. Ryu and D. Han, Biochem. Biophys. Res. Commun., 2014, 448, 114–119 CrossRef CAS PubMed.
- C. Renner, J. Piehler and T. Schrader, J. Am. Chem. Soc., 2006, 128, 620–628 CrossRef CAS PubMed.
- S. H. Lee, Y. Hoshino, A. Randall, Z. Y. Zeng, P. Baldi, R. A. Doong and K. J. Shea, J. Am. Chem. Soc., 2012, 134, 15765–15772 CrossRef CAS PubMed.
- C. Armutcu, N. Bereli, E. Bayram, L. Uzun, R. Say and A. Denizli, Colloids Surf., B, 2014, 114, 67–74 CrossRef CAS PubMed.
- Y. J. Chen, Z. C. Xiong, L. Y. Zhang, J. Y. Zhao, Q. Q. Zhang, L. Peng, W. B. Zhang, M. L. Ye and H. F. Zou, Nanoscale, 2015, 7, 3100–3108 RSC.
- H. R. Nodeh, W. A. W. Ibrahim, M. A. Kamboh and M. M. Sanagi, RSC Adv., 2015, 5, 76424–76434 RSC.
- N. Griffete, J. Fresnais, A. Espinosa, C. Wilhelm, A. Bee and C. Menager, Nanoscale, 2015, 7, 18891–18896 RSC.
- H. Deng, X. L. Li, Q. Peng, X. Wang, J. P. Chen and Y. D. Li, Angew. Chem., Int. Ed., 2005, 44, 2782–2785 CrossRef CAS PubMed.
- M. E. Çorman, C. Armutcu, L. Uzun, R. Say and A. Denizli, Colloids Surf., B, 2014, 123, 831–837 CrossRef PubMed.
- W. Qin and Y. Gao, Chin. J. Biotechnol., 2015, 31, 1387–1392 Search PubMed.
- Y. Hu, X. H. Gao, L. Yu, Y. R. Wang, J. Q. Ning, S. J. Xu and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 5636–5639 CrossRef CAS PubMed.
- L. Korosi, M. Prato, A. Scarpellini, J. Kovacs, D. Domotor, T. Kovacs and S. Papp, Appl. Surf. Sci., 2016, 365, 171–179 CrossRef CAS.
- H. Nourolahi, M. A. Bolorizadeh and A. Behjat, Vacuum, 2016, 123, 29–34 CrossRef CAS.
- J. B. Zhu, Y. L. Xu, J. Wang, J. P. Wang, Y. Bai and X. F. Du, Phys. Chem. Chem. Phys., 2015, 17, 19885–19894 RSC.
- E. T. Salim, Y. Al-Douri, M. S. Al Wazny and M. A. Fakhri, Solar Energy, 2014, 107, 523–529 CrossRef CAS.
- S. J. Yan, L. N. Wang, T. H. Wang, L. Q. Zhang, Y. F. Li and S. L. Dai, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 6 CrossRef.
- V. Iannotti, S. Amoruso, G. Ausanio, A. C. Barone, C. Campana, X. Wang and L. Lanotte, Appl. Surf. Sci., 2009, 255, 5224–5227 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The detailed synthetic procedure, graphics of 1H NMR, TGA, XRD and ITC, and optimisation of CE. See DOI: 10.1039/c6ra24560a |
| This journal is © The Royal Society of Chemistry 2016 |