
A comparative study of the crystalline structure and mechanical properties of carbon fiber/polyamide 6 composites enhanced with/without silane treatment
Lin Sanga,
YuKai Wanga,
Guangyi Chena,
Jicai Lianga and
Zhiyong Wei *b
*b
aSchool of Automotive Engineering, Dalian University of Technology, Dalian 116024, China
bDepartment of Polymer Science and Materials, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, China. E-mail: zywei@dlut.edu.cn
First published on 4th November 2016
Abstract
Carbon fiber (CF) reinforced polyamide 6 (PA6) composites were manufactured by extrusion compounding and injection molding. The carbon fiber was considered in two forms including untreated and treated with silane coupling agent. The main focus of this study was to investigate the effects of the silane treatment on the mechanical properties and crystalline structure of the composites. Mechanical test results showed that the tensile, impact and flexural strength were significantly increased by incorporation of silane-treated carbon fiber. In particular, the specific tensile, impact and flexural strength of silane-treated CF composites with 20% fibre mass fraction are, respectively, 42%, 51.6% and 30% higher than those of untreated CF composites. Scanning electron microscopy examination showed that the tensile fracture surface of silane-treated CF composites failed in a fiber breakage pattern while the untreated CF composites fractured via a fiber pull-out pattern, suggesting an enhanced interfacial adhesion with the matrix. In addition, the incorporation of silane-treated CF increased the degree of crystallinity, promoted the formation of the thermodynamic crystalline form and induced the specific transcrystalline structure, as indicated by differential scanning calorimetry (DSC), X-ray diffraction (XRD) and polarizing optical microscopy (POM) analysis. Moreover, the mechanism of silane-treated CF reinforced PA6 composites with improved mechanical properties was discussed.
1. Introduction
Currently, thermoplastic and thermosetting materials as matrices dominate the area of fiber reinforced polymer composites for vital engineering applications such as aerospace, aircraft or automotive industries. However, with increasing environmental awareness and enforcing legislations, short fiber reinforced thermoplastic materials have received increasing interest because of their advantages such as lightweight, renewability and the expected thermo/mechanical performances.1–3 Among the thermoplastic candidates, polyamide 6 (PA6), as a semicrystalline polymer, has become a strong competitor matrix with remarkable mechanical properties and versatile processibility.4 These advantages make PA6 an ideal matrix material especially for fiber-reinforced composites.Carbon fiber (CF), owning remarkably high specific strength and modulus, electrical and thermal properties, has been considered an excellent candidate reinforcement for thermoplastic materials.5–7 However, despite the excellent mechanical characteristics, bare CF is known to suffer from poor adhesion to matrix polymers.8–10 Hence, various efforts have been made on the surface of the fiber, i.e., chemical modification,11 polymer grafting on the surface of the fibers, incorporation of compatibilizer, such as maleated polymer12,13 or treatment with coupling agents.14 These treatments would promote proper interfacial adhesion and ensure an efficient stress transferring from thermoplastic matrix to fibers.15–17 Of the above methods, silane treatment is widely used for the surface modification of fiber or inorganic filler, establishing efficient interfacial bonding between fiber and polymeric matrix. Among various coupling agents, organo-silane (R–Si–(OR′)3) is the most effective, commercially available, and low cost product.18,19
Using different silane coupling agent, Jiang et al. showed that surface silanization of carbon fiber improved tensile strength of CF/PU composites by 18.3%, compared with untreated fibers.20 Kim et al. modified carbon fiber with an isocyanato silane compound, and both the tensile strength and thermal stability of silicone rubber/carbon fiber were improved.21 While most of these works were in agreement with improved interfacial and mechanical properties after silane treatment, there were very limited researches regarding the internal mechanism of the evolution in mechanical properties. For crystalline or semi-crystalline thermoplastic polymers (i.e. PLA, PBS, PA6), the thermoplastic intrinsic morphological features such as crystal size, nucleus density, crystallinity and transcrystallization also have profound effects on the ultimate properties of carbon fiber reinforced thermoplastic composites.22,23 However, few works are focused on the effects of silane-treated fibers on the crystalline structure and thermal/mechanical properties from micro- and macro-view.24–26
The aim of present study is to investigate the effects of silane-treated carbon fiber on the crystalline structure, mechanical properties of the semi-crystalline polyamide-based composites, and clarify the relationship between the crystalline microstructure and macroscopic mechanical performance of the composites. The silane-treated CF reinforced PA6 composites were prepared by extrusion compounding and injection molding technique. The degree of crystallinity and crystalline structure are examined and observed by differential scanning calorimetry (DSC), X-ray diffraction (XRD) and polarizing optical microscopy (POM), and the macroscopic mechanical performance are comparatively evaluated by tensile test, impact test, and three-point bending test.
2. Experimental
2.1 Materials
PA6 used in this study was an injection-molding-grade resin, supplied from Yueyang Baling Jiayun Petrochemical. The melt flow index (MFI) of 2.4–3.6 g 10 min−1. PA6 was vacuum dried at 100 °C for 12 h prior to use. Carbon fiber tow (PAN (polyacrylonitrile)-based carbon fiber, 3k) was provided by Dalian Xingke Carbon Fiber Co., Ltd., LiaoNing Province, China. The characteristics of the CF are as follows: the diameter 7 μm, the density is 1.78 g cm−3, the tensile strength >3.3 GPa, and the tensile modulus 230–260 GPa. 3-Aminopropyltriethoxy silane coupling agent (KH550, NH2CH2CH2CH2Si(OC2H5)3, 97% purity), acetone (99.5% purity), alcohol (95% purity) were purchased from J&K Scientific Ltd., China.2.2 Preparation of composites
3-Aminopropyltriethoxy silane coupling agent (KH550) was used to modify the carbon fiber surface. The modification of carbon fibers was performed as follows: the CF was firstly immersed in acetone/ethanol for 2 h to clean and polarize the fiber surfaces. After washed to neutrality with deionized water, the CF was soaked into the mixture solution of KH550 with certain ratio of acetone/water solution being solvent at a concentration of 0.75 wt% for 30 min. Then, the modified carbon fiber (MCF) was baked under vacuum at 120 °C for 2 h.The raw PA6 was dried at 80 °C for 12 h to before blending. Carbon fiber reinforced PA6 composites were prepared by melting mixing PA6 with untreated, silane-treated CF in a twin screw extruder (SHJ20-X40, D = 20 mm, L/D = 40, Nanjing Giant Machinery Co., Ltd. China). PA6 was fed from the hopper, and the continuous carbon fiber was automatically fed through the fiber feeding port by rotation of the screw. The CF was sheared to be short and dispersed in PA6 matrix due to the shear force during extruding. The temperatures along the barrel from feeding zone to die were set at 230, 235, 240, 245 and 240 °C, respectively. The melting extrudate was cooled in a water bath and subsequently pelletized. The resulted pellets were dried under 85 °C for at least 12 h before injection molding. Composite specimens reinforced with untreated CF and silane-treated CF were marked with the codes of CF/PA6 and MCF/PA6, respectively. Furthermore, composites containing 10 wt% and 20 wt% fibers were coded with CF10/PA6, CF20/PA6, MCF10/PA6 and MCF20/PA6, respectively.
Standard specimens used for tensile, impact and flexural tests were prepared on an injection molding machine (XTK 1200, Xiatian General Machinery, Ningbo, China). Neat PA6 was set as control samples. Composition of composites was listed in Table 1.
| Sample | CF loading (wt%) | MCF loading (wt%) | Tensile strength (MPa) | Strain at break (%) | Tensile modulus (MPa) |
|---|---|---|---|---|---|
| PA6 | 0 | 0 | 78.4 ± 3.1 | 300 ± 10.5 | 26.2 ± 1.9 |
| CF10/PA6 | 10 | — | 107.4 ± 2.9 | 5.0 ± 1.0 | 2249.6 ± 507.9 |
| CF20/PA6 | 20 | — | 124.6 ± 3.8 | 4.2 ± 0.7 | 3067 ± 601.7 |
| MCF10/PA6 | — | 10 | 130.7 ± 3.7 | 7.5 ± 0.6 | 1757.9 ± 190 |
| MCF20/PA6 | — | 20 | 176.2 ± 5.4 | 5.0 ± 1.2 | 3766.9 ± 1012.1 |
2.3 Characterization
| Samples | CF loading (wt%) | MCF loading (wt%) | Flexural strength (MPa) | Strain at break (%) |
|---|---|---|---|---|
| PA6 | 0 | 0 | 70.3 ± 5.3 | 16.1 ± 3.8 |
| CF10/PA6 | 10 | — | 84.9 ± 4.6 | 1.5 ± 0.2 |
| CF20/PA6 | 20 | — | 107.5 ± 3.5 | 1.9 ± 0.7 |
| MCF10/PA6 | — | 10 | 115.5 ± 7.8 | 5.0 ± 0.6 |
| MCF20/PA6 | — | 20 | 134.9 ± 4.4 | 3.7 ± 0.4 |
The dumbbell samples were conducted according to GB/T 1040:2-2006. At least five specimens were tested for each set of samples and the average results were reported.
The unnotched impact tests of the samples were carried out with XQZ-II impact testing machine (JJ-Test Chengde, Hebei, China). The samples of 50 mm × 6 mm × 4 mm in size were tested at room temperature according to GB/T 1843-2008. An average value on five tests was taken for each material.
 | (1) |
3. Results and discussion
3.1 Surface characteristics and silane treatment of MCF
The schematic representation of coupling mechanism of silane treatment was illustrated in Fig. 1. A commercially available organo-silane, 3-aminopropyltriethoxy (KH550), was adopted in this study. During silane treatment, the ethoxy groups (–OC2H5) were firstly hydrolyzed and thus formed hydroxyl groups in reactive silanol, and then react with hydroxyl groups in basalt fiber surface.18,19 Furthermore, the –NH2 groups, as functional groups, were responsible for establishing covalent bonds with the –COOH end groups of PA6, which was expected to establish the interfacial bonding between the reinforcing element (CF) and the polymer matrix (PA6). | ||
| Fig. 1 Schematic steps of silane treatment with carbon fiber composites. | ||
The surface morphologies of untreated CF and silane-treated CF were observed by SEM (Fig. 2). As shown in Fig. 2a, the surface of CF exhibited a clean and smooth surface. On the other hand, after silane treatment, a rough surface with small granules was found in MCF, which can be attributed to the deposition of the silane layer on the surface of fibers.
 | ||
| Fig. 2 SEM images of (a) CF and (b) MCF samples. | ||
3.2 Mechanical properties of composites
Fig. 3 showed the standard tensile specimens and typical stress–strain curves for PA6, CF/PA6 and MCF/PA6, which gave an intuitive trend of mechanical properties with varying fiber loading or fiber pre-treatment. The Young's modulus, tensile strength and elongation at break extracted from the stress–strain curves were summarized in Table 1. For the neat PA6, the matrix exhibited a yielding behavior and significantly high elongation at break, which are typical characteristics for semi-crystalline ductile polymers. However, compared with neat PA6, all the composite samples showed almost a linear tensile behavior without any cold flowing and the elongation at break began to decrease dramatically, with a failure strain between 4.2% and 7.5%, which indicated that the addition of carbon fiber interfered the mobility or deformability of the matrix. With increasing fiber loading, the tensile strength and elastic modulus were greatly improved, owing to the excellent mechanical properties of carbon fiber. Moreover, a further increase was observed in the MCF/PA6. In the case of MCF20/PA6, the tensile strength and tensile modulus were up to the maximum value point of 176.2 MPa and 3766.9 MPa, respectively, which were about 125% higher than those of neat PA6 at 78.4 ± 3.1 MPa. Mylsamy and Rajendran28 indicated that the tensile properties in composites depend mainly on the fiber orientation and the adhesion between the fibers and matrix. In this work, as the carbon fibers are randomly oriented in the composites, the tensile strength and elastic modulus would be strongly affected by the quality of the interphase of the carbon fiber-reinforced PA6 composites.29 Therefore, the silane coupling agent supplied a major bonding between fiber and matrix, which made MCF/PA6 composite undertake more stress loading and more energy during crack propagation.28,30,31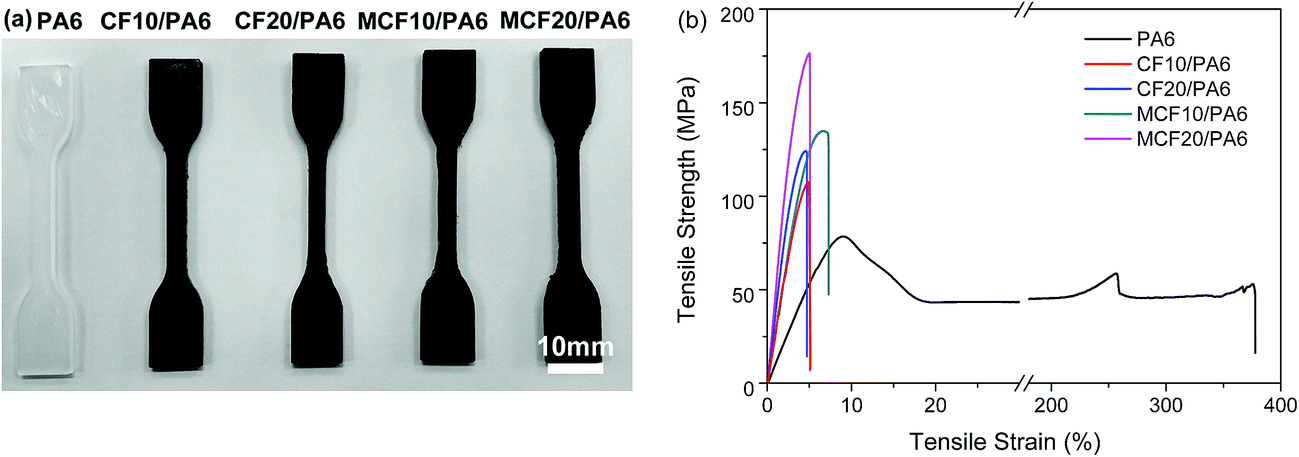 | ||
| Fig. 3 (a) Standard specimens for tensile test, (b) tensile stress–strain behavior of PA6, CF/PA6 and MCF/PA6. | ||
A close inspection at the tensile failure surface was observed by SEM (Fig. 4). It can be observed that neat PA6 matrix endured a plastic deformation before complete fracture. In the case of untreated CF/PA6 composites, plenty of untreated carbon fibers were lip through the matrix, and big interspaces between CF and PA6 were obviously visible, leading to the failure of composites through a fiber/matrix interfacial debonding and fiber pull-out pattern (Fig. 4b and d). This can be attributed to the poor interaction between the fiber and matrix.32,33 On the other hand, for silane-treated composites (MCF/PA6), an amount of resin was observed adhering to the MCF surface. Meanwhile, few cavities and voids were observed, indicating the silane-treated composites failed in a dominant pattern of matrix cracking and fiber breakage (Fig. 4c and e). These results implied a strong interaction between the silane-treated fibers and the matrix, which was consistent with the results of the tensile properties.
 | ||
| Fig. 4 SEM micrographs of tensile fracture surfaces of (a) PA6, (b) CF10/PA6, (c) MCF10/PA6, (d) CF20/PA6 and (e) MCF20/PA6. | ||
The impact properties of PA6, CF/PA6 and MCF/PA6 were studied by unnotched Izod impact test, which was directly correlated to the composite toughness under stress at a high speed. As shown in Fig. 5, the impact strength for neat PA6 was 7.5 ± 0.6 kJ m−1; the incorporation of fiber improved the impact strength to some extent. However, the impact strength did not shown much difference in varying content of fiber loading. For CF10/PA6 and CF20/PA6, the impact strength were 11.3 ± 0.5 and 12.2 ± 0.2 kJ m−1 respectively, which was improved by about 60% compared to neat PA6. After silane treatment, the maximum value at 18.5 ± 0.6 kJ m−1 of the unnotched impact strength was obtained from the MCF20/PA6, which was 52% higher than untreated CF20/PA6. Consequently, silane treatment is beneficial for the increase of the impact strength of the composites.
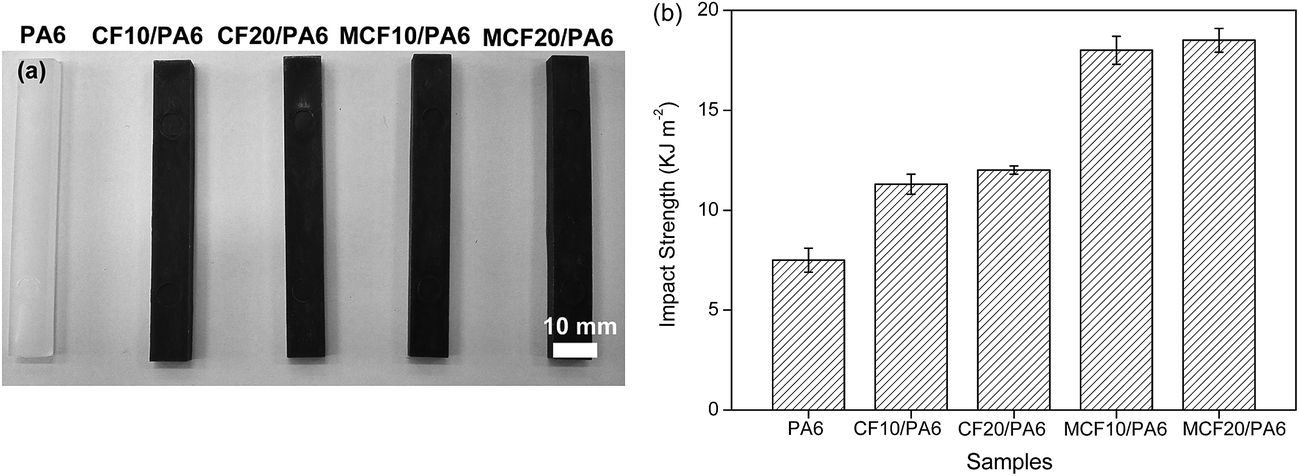 | ||
| Fig. 5 (a) Standard specimens for impact test, (b) unnotched Izod impact strength of PA6, CF/PA6 and MCF/PA6. | ||
Fig. 6 showed the stress–strain for PA6, CF/PA6 and MCF/PA6 under bending. It can be observed that a significant improvement of flexural strength was achieved with addition of carbon fibers, and this trend was further increased with the addition of silane-treated CF. CF10/PA6 and CF20/PA6 specimens with untreated CF failed at an average bending stress at 84.9 and 107.5 MPa, respectively, which were higher than that of neat PA6 at 70.3 MPa. On the other hand, the MCF10/PA6 and MCF20/PA6 specimens with silane-treated CF failed at a bending stress of 107.5 MPa and 134.9 MPa. Similar to the tensile strength and impact strength, it is believed that the high strength of CF and improved interfacial interaction may synergistically make the composite undertake more test stress.34
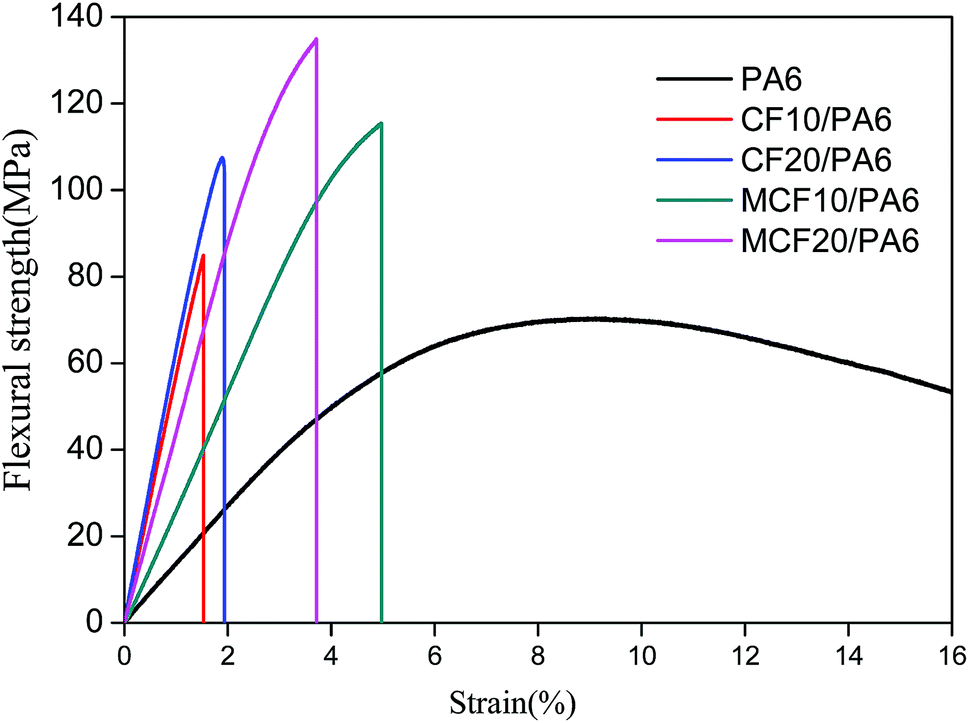 | ||
| Fig. 6 Flexible strength of PA6, CF/PA6 and MCF/PA6. | ||
3.3 Crystallization behavior
PA6 is a semi-crystalline polymer, and its degree of crystallinity and crystalline morphology are directly correlated with the physical properties. It is an important issue to investigate the influence of untreated CF and silane-treated CF on the crystallization behaviors of PA6, since the crystallinity of PA6 matrix plays an important role in the physical properties and processability for the composites. Fig. 7 illustrated the variation trend of crystallization temperature at peak (Tp) and crystallization enthalpy (ΔHc) at different cooling rates (Φ) via DSC measurements. All the data derived from non-isothermal crystallization were summarized in Table 3. Compared with the neat PA6, the Tps of the composites shifted to high temperatures, accompanied by an increase in ΔHcs under increasing cooling rates. Based on the data from Table 3, by varying cooling rate (5–20 °C min−1), the Xc of PA6 was varied from 21.7 to 23.2%, while the Xc of CF/PA6 composites were varied from 25.6 to 32.9%, and that of MCF/PA6 were varied from 33.0 to 42.7%. These results indicated that the incorporation of fiber reinforcement significantly enhanced the crystallinity of PA6 under non-isothermal condition. Both ΔHcs and Xc of MCF/PA6 showed a distinct increase compared with CF/PA6 suggesting that the incorporation of silane-treated CF facilitated the crystallization of PA6 and greatly enhanced the degree of crystallinity of PA6.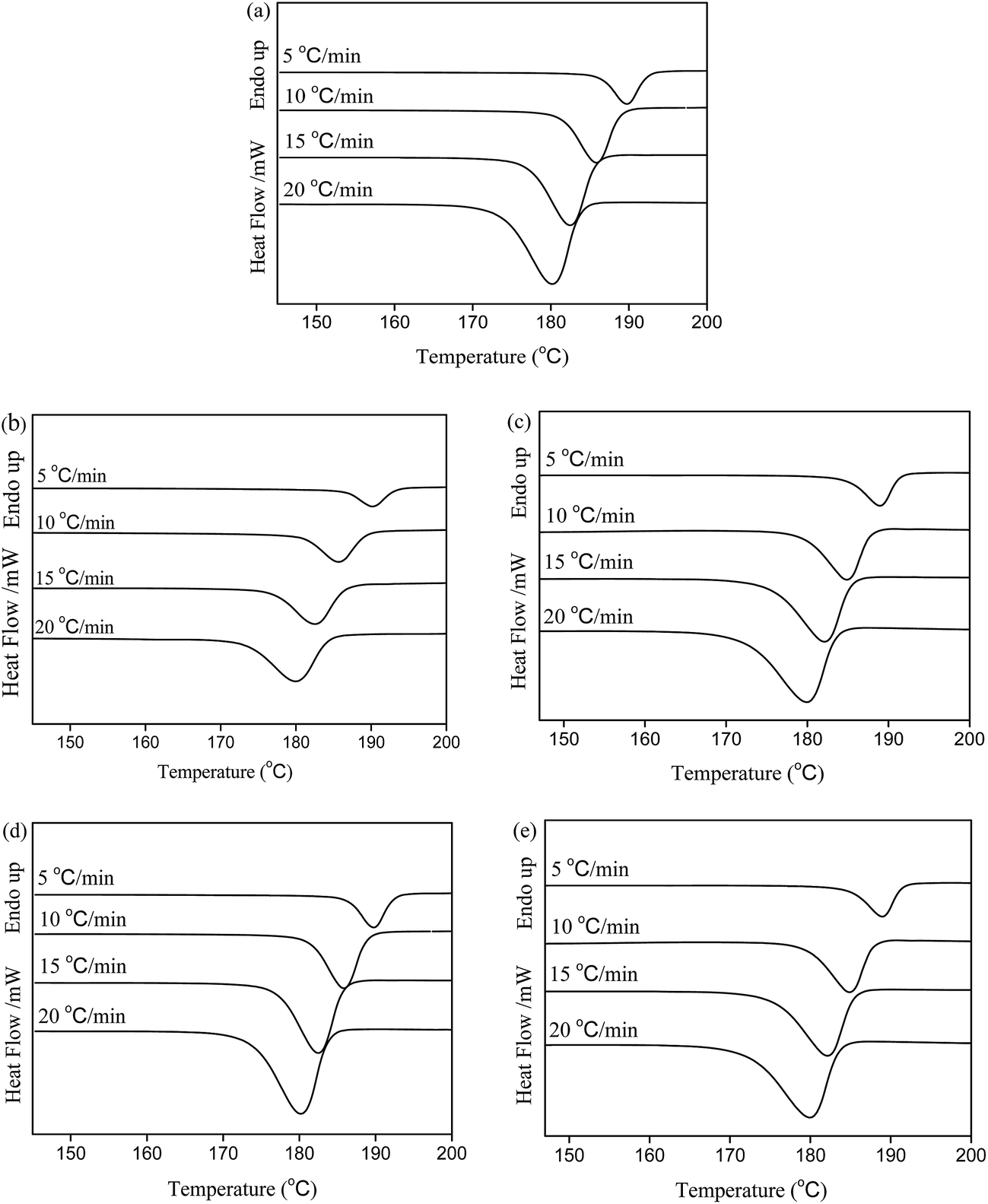 | ||
| Fig. 7 Non-isothermal crystalline curves of (a) PA6, (b) CF10/PA6, (c) CF20/PA6, (d) MCF10/PA6 and (e) MCF20/PA6 at varying cooling rates. | ||
| Samples | Cooling rate (°C min−1) | Tp (°C) | Normalized ΔHc (J g−1) | Xc (%) |
|---|---|---|---|---|
| PA6 | 5 | 185.2 | 41.4 | 21.7 |
| 10 | 179.6 | 42.3 | 22.2 | |
| 15 | 175.9 | 43.3 | 22.7 | |
| 20 | 173.4 | 44.3 | 23.2 | |
| CF10/PA6 | 5 | 189.8 | 44.0 | 25.6 |
| 10 | 185.9 | 44.1 | 25.7 | |
| 15 | 182.4 | 44.8 | 26.1 | |
| 20 | 180.2 | 44.2 | 25.7 | |
| CF20/PA6 | 5 | 189.4 | 45.1 | 29.5 |
| 10 | 184.1 | 46.6 | 30.5 | |
| 15 | 180.5 | 48.2 | 31.6 | |
| 20 | 178.2 | 50.2 | 32.9 | |
| MCF10/PA6 | 5 | 189.8 | 67.2 | 39.1 |
| 10 | 185.8 | 69.6 | 40.5 | |
| 15 | 182.4 | 70.2 | 40.9 | |
| 20 | 180.1 | 73.4 | 42.7 | |
| MCF20/PA6 | 5 | 188.9 | 50.4 | 33.0 |
| 10 | 184.9 | 55.2 | 36.1 | |
| 15 | 182.1 | 55.9 | 36.6 | |
| 20 | 179.8 | 58.0 | 38.0 |
3.4 Crystal structure and spherulitic morphology
The effect of untreated CF and silane-treated CF on crystalline structure was firstly studied by XRD analysis. Fig. 8 showed XRD patterns of the PA6, CF/PA6 and MCF/PA6 samples. There are several kinds of crystalline forms of PA6, and α and γ are the frequently observed forms of PA6 crystalline.35 As shown in Fig. 8a, the crystalline structure of neat PA6 matrix was dominated by γ form crystal phase, which was characterized with an intense peak at 22° (100 reflection) and a weak shoulder at 23° (01 + 200 reflections).36 On the other hand, CF/PA6 and MCF/PA6 mainly crystallized into the thermodynamically stable α form crystal phase. The α form showed two characteristic peaks at 21° (200 reflection) and 24° (002 + 202 reflections). The XRD results indicated that the reinforcement of CF would induce the crystallization into α form. Some published work has proved that the γ form crystalline fracture was more ductile than α for crystalline fracture.37 This might be considered as another reason that PA6 matrix exhibited a ductile tensile stress–strain curve. | ||
| Fig. 8 XRD patterns of PA6, CF/PA6 and MCF/PA6. | ||
Fig. 9 displayed typical polarized optimal micrographs of crystalline morphology of PA6 in the presence of CF or MCF with different fiber loading after isothermal crystallization at 213 °C for 1 h. Generally, the presence of fiber has an important influence on the crystalline morphology of PA6. As shown in Fig. 9a, the PA6 matrix exhibited fine and well-dispersed spherulites. After the incorporation of CF, the spherulitic size of PA6 dramatically decreased. It is deduced that the incorporation of fibers performed a heterogeneous nucleating effect, thus enhanced the crystallinity of PA6. Remarkably, for MCF/PA6, it is interesting to find that some crystalline were grown surrounding the surface of silane-treated CF (Fig. 9c and e). This special oriented interfacial microstructure is identified as transcrystals (TCs).38 Instead, no TC structure was found on the surface of untreated CF. This phenomenon may be due to the fact that the smooth and inert surface of untreated carbon fiber. Clearly, the addition of silane-treated CF largely improved the nucleation ability of PA6 and increased the occurrence of transcrystallization.
 | ||
| Fig. 9 Polarized optical micrographs of spherulitic morphology of (a) neat PA6, (b) CF10/PA6, (c) MCF10/PA6, (d) CF20/PA6 and (e) MCF20/PA6. | ||
Fig. 10 highlighted the schematic representation of PA6 (trans-)crystallization at the presence of untreated CF and silane-treated CF (MCF). Initially, the PA6 melt surround fiber surface. Due to the efficient interaction between silane-treated CF surface and the PA6, the molecular chains of PA6 formed clusters on the surface of MCF, forming a number of nucleation sites of the MCF. Subsequently, these lamellae began to grow perpendicular to the fiber surface. The difference between the spherulites and transcrystallization is that the spherulites start to grow rapidly from the nuclei in a three dimensional way, while the transcrystallizations grow from the nuclei in a one- or two-dimensional way.
 | ||
| Fig. 10 Schematic illustrations of the composites of PA6 and CF or MCF, and mechanisms for better interfaces after silane treatment. | ||
From the results of XRD, DSC and POM images, our observations demonstrated that the silane-treated CF promoted the formation of transcrystals, increased the crystallinity and accelerated transcrystallization. In general, polymer transcrystallization in the presence of the fiber is a complex process and the formation mechanism has not been fully understood. In our work, the topography and surface group of the fibers, together with interfacial interaction played important roles in controlling polymer crystallization. Firstly, the rough and granular microstructures (evidenced by Fig. 2b) of MCF facilitated high-density heterogeneous nucleation sites of transcrystals than the untreated CF (Fig. 10c), which has been proved by previous literatures.39 Secondly, the MCF possessed plenty of hydroxyl and amino groups after silane treatment, the efficient interaction between fiber and matrix promoted the formation of transcrystals.40 On the contrary, the untreated CF induced relatively low-density heterogeneous nucleation sites because of its smooth and inert surface (Fig. 10b). This may also explained the relatively high crystallinity of the MCF/PA6 compared with neat PA6 and PA/CF. Besides the interaction between the MCF and PA6 matrix, it is believed that the transcrystallinity can further improve the adhesion of the fiber to the polymer matrix.41,42 Liu et al. showed that the presence of silane-treated basalt fiber, glass fiber and carbon fiber can induce the TC structure and improved the compatibility between the fiber and PLA.39 Zhou et al. demonstrated that high density of TC structure greatly contributed to improving the interfacial adhesion and mechanical properties of PBS/ramie fiber composites.23 Therefore, in our work, it is believed that MCF/PA6 with an ordered interfacial transcrystalline structure can bear larger stress before its failure and lead to higher load transfer efficiency than CF/PA6. Consequently, the enhanced mechanical properties of MCF/PA6 were ascribed to three important factors, including the reinforcement of CF, the high degree of crystallinity and transcrystalline microstructure of PA6 matrix.
3.5 Thermal stability
The thermal stabilities of PA6, CF/PA6 and MCF/PA6 were evaluated by TGA with a temperature range from room temperature to 550 °C, which revealed their behaviors of thermal degradation (Fig. 11). From the weight loss profiles, neat PA6, CF/PA6 and MCF/PA6 occurred through typical one-step degradation. The neat PA6 matrix underwent an initial decomposition with 5 wt% of weight loss at a temperature of around 405 °C and then a rapid decomposition with a maximum weight loss at around 500 °C due to the chain scission, demonstrating a good thermal stability of PA6, whereas neat PA6 almost full pyrolysis with only 0.85 wt% of residual char (Table 4). It is interestingly noticed that the CF/PA6 and MCF/PA6 showed a slightly higher decomposition temperature with 50 wt% and maximum weight loss than neat PA6, suggesting that improved thermal stability after the incorporation of carbon fiber. It was reported that the incorporation of carbon materials into polymeric matrix decreased the heat release rate, therefore they retard the decomposition to some extent.43 | ||
| Fig. 11 TGA curves of PA6, CF/PA6 and MCF/PA6 composites. | ||
| Sample | T5 (°C) | T50 (°C) | Tmax (°C) | Residue (%) (at 600 °C) |
|---|---|---|---|---|
| PA6 | 406 | 461 | 467 | 0.85 |
| CF10/PA6 | 399 | 466 | 471 | 11.1 |
| CF20/PA6 | 403 | 469 | 470 | 20.2 |
| MCF10/PA6 | 418 | 468 | 473 | 10.0 |
| MCF20/PA6 | 401 | 467 | 471 | 19.2 |
4. Conclusion
The aim of this study is to investigate the effects on silane-treated carbon fiber on the crystalline structure and mechanical properties of fiber-reinforced PA6 composites, exploring the relationship between microstructure and macroscopic performance. According to the mechanical test, the tensile strength, impact strength and flexural strength were greatly increased for silane-treated CF composites in comparison with untreated CF composites. The morphology of tensile fracture surface demonstrated that MCF/PA6 specimen failed through matrix cracking and fiber breakage, while the untreated CF/PA6 specimen fractured via a pull-out pattern. These mechanical results confirmed that the silane treatment of carbon fibers lead to strong interfacial bond between the fiber and the matrix. From the micro view of MCF/PA6, the silane-treated CF acted as heterogeneous nucleation effect, promoting the transformation from γ into thermodynamically stable α form with high crystallinity. Moreover, the transcrystalline structure was observed, which further improve the interfacial adhesion between fiber and matrix. Consequently, silane treatment of carbon fiber is an effective alternative to contribute an evolution of mechanical properties.Acknowledgements
This work was financially supported by the Fundamental Research Funds for the Central Universities (DUT15RC(3)036).References
- C. S. Qia, V. K. Yadama, Q. Guo and M. P. Wolcott, Ind. Crops Prod., 2013, 45, 455–460 CrossRef
.
- F. Sliwa, N. El Bounia, F. Charrier, G. Marin and F. Malet, Compos. Sci. Technol., 2012, 72, 1733–1740 CrossRef CAS
- R. Malkapuram, V. Kumar and S. N. Yuvraj, J. Reinf. Plast. Compos., 2009, 28(10), 1169–1189 CrossRef CAS
- E. C. Botelho, L. Figiel, M. C. Rezende and B. Lauke, Compos. Sci. Technol., 2003, 63(13), 1843–1855 CrossRef CAS
- Y. X. He, Q. Li, T. Kuila, N. H. Kim, T. W. Jiang, K. T. Lau and J. H. Lee, Composites, Part B, 2013, 44, 533–539 CrossRef CAS
- M. H. Al-Saleha and U. Sundararaj, Composites, Part A, 2011, 42, 2126–2142 CrossRef
- F. Rezaei, R. Yunus and N. A. Ibrahim, Mater. Des., 2009, 30, 260–263 CrossRef CAS
- X. Li, L. G. Tabil and S. Panigrahi, J. Polym. Environ., 2007, 15, 25–33 CrossRef
- T. J. Keener, R. K. Stuart and T. K. Brown, Composites, Part A, 2004, 35, 357–362 CrossRef
- A. Arbelaiz, B. Fernandez, G. Cantero, R. Llano-Ponte and A. Valea, Composites, Part A, 2005, 36, 1637–1644 CrossRef
- G. A. Valadez, U. Cervantes, R. Olayo and P. Herrera-Franco, Composites, Part B, 1999, 30, 321–331 CrossRef
- X. Q. Zhang, H. B. Xu and X. Y. Fan, RSC Adv., 2014, 4, 12198–12205 RSC
- A. Arbelaiz, B. Fernandez, J. A. Ramos, A. Retegi, R. Llano-Ponte and I. Mondragon, Compos. Sci. Technol., 2005, 65, 1582–1592 CrossRef CAS
- R. Agrawal, N. S. Saxena, K. B. Sharma, S. Thomas and M. S. Sreekala, Mater. Sci. Eng., 2000, 277, 77–82 CrossRef
- E. K. Drown, H. Almoussawi and L. T. Drzal, J. Adhes. Sci. Technol., 1991, 5(10), 865–881 CrossRef CAS
- G. R. Baran, S. Debnath, S. L. Wunder and J. I. McCool, Dent. Mater., 2003, 19(5), 441–448 CrossRef
- J. L. Thomason and L. J. Adzima, Composites, Part A, 2001, 32(3–4), 313–321 CrossRef
- T. Deak, T. Gzigany, P. Tamas and C. Nemeth, eXPRESS Polym. Lett., 2010, 4(10), 590–598 CrossRef CAS
- J. M. He and Y. D. Huang, J. Appl. Polym. Sci., 2007, 106, 2231–2237 CrossRef CAS
- S. Jiang, Q. F. Li, Y. H. Zhao, J. W. Wang and M. Q. Kang, Compos. Sci. Technol., 2015, 110, 87–94 CrossRef CAS
- E. S. Kim, T. H. Lee, E. J. Kim and J. S. Yoon, J. Appl. Polym. Sci., 2012, 126(SI), 410–418 CrossRef
- S. L. Gao and J. K. Kim, Composites, Part A, 2000, 31, 517–530 CrossRef
- M. Zhou, J. J. Yan, Y. H. Li, C. Z. Geng, C. He, K. Wang and Q. Fu, RSC Adv., 2013, 3, 26418–26426 RSC
- M. D. Samper, R. Petrucci, L. Sánchez-Nacher, R. Balart and J. M. Kenny, Polym. Compos., 2015, 36, 1205–1212 CrossRef CAS
- A. Orue, A. Jauregi, U. Unsuain, J. Labidi, A. Eceiza and A. Arbelaiz, Composites, Part A, 2016, 84, 186–195 CrossRef CAS
- M. T. Zafar, S. N. Maiti and A. K. Ghosh, RSC Adv., 2016, 6, 73373–73382 RSC
- I. Campoy, M. A. Gomez and C. Marco, Polymer, 1998, 39, 6279–6288 CrossRef CAS
- K. Mylsamy and I. Rajendran, Mater. Des., 2011, 32, 4629–4640 CrossRef CAS
- M. Kodal, Z. D. Topuk and G. Ozkoc, J. Appl. Polym. Sci., 2015, 132(48), 42564 CrossRef
- Z. Y. Liu, B. Hao and Y. G. Zhang, RSC Adv., 2015, 5, 40668–40677 RSC
- H. Y. Han, X. D. Wang and D. Z. Wu, Composites, Part A, 2012, 43, 1947–1958 CrossRef CAS
- J. Li and C. L. Cai, Curr. Appl. Phys., 2011, 11, 50–54 CrossRef
- N. G. Karsli and A. Aytac, Composites, Part B, 2013, 51, 270–275 CrossRef CAS
- T. J. Keener, R. K. Stuart and T. K. Brown, Composites, Part A, 2004, 35(3), 357–362 CrossRef
- X. L. Yan, Y. Imain, D. Shimamoto and Y. J. Hotta, Polymer, 2014, 55, 6186–6194 CrossRef CAS
- P. Bernadó, C. Alemán and J. Puiggalí, Eur. Polym. J., 1999, 35, 835–847 CrossRef
- M. Ito, K. Mizuochi and T. Kanamato, Polymer, 1998, 39, 4593–4598 CrossRef CAS
- H. Quan, Z. M. Li, M. B. Yang and R. Huang, Compos. Sci. Technol., 2005, 65, 999–1021 CrossRef CAS
- T. Liu, X. J. Yu, F. M. Xu, X. L. Zhao, A. Lu, J. H. Wang, X. Z. Wang and T. L. Liu, Polym.–Plast. Technol. Eng., 2012, 51, 597–604 CrossRef CAS
- J. P. Abdou, K. J. Reynolds, M. R. Pfau, J. van Staden, G. A. Braggin, N. Tajaddod, M. Minus, V. Regurero, J. J. Vilatela and S. Zhang, Polymer, 2016, 91, 136–145 CrossRef CAS
- P. V. Joseph, K. Joseph, S. Thomas, C. K. S. Pillai, V. S. Prasad and G. Groeninckx, Composites, Part A, 2003, 34, 253–266 CrossRef
- Y. L. Mi, X. Y. Chen and Q. P. Guo, J. Appl. Polym. Sci., 1997, 64, 1267–1273 CrossRef CAS
- X. Wen, Y. Wang, J. Gong, J. Liu, N. Tian, Y. Wang, Y. H. Wang, Z. W. Jiang, J. Qiu and T. Tang, Polym. Degrad. Stab., 2012, 97(5), 793–801 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2016 |