
Efficient drug delivery system for bone repair by tuning the surface of hydroxyapatite particles
Abstract
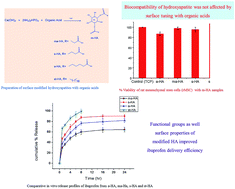
A limited blood flow to skeletal tissues results in minimal therapeutic effect of drugs being administered to a patient using conventional ways. To obtain sufficient amount of drug at an effected site, implanted drug delivery systems based on biomaterials can be used. In this study, surface modified hydroxyapatites (m-HA) were prepared and evaluated as drug delivery systems. The effect of modifiers on surface properties of HA and their in vitro drug delivery efficiency were investigated. For synthesis of m-HA, a simple in situ co-precipitation method was used. Hydroxyapatite was subjected to surface modification by various carboxylic acids such as adipic acid, malonic acid, succinic acid and stearic acid. This surface modification affected its surface properties such as surface area, pore size, pore volume, particle size and crystallinity. The m-HA were characterized by Fourier transform infrared spectroscopy (FT-IR), X-ray diffraction (XRD) and thermogravimetric analysis (TGA). Brunauer–Emmett–Teller (BET) technique was used to compute surface properties of m-HA. The highest BET surface area of 143 m2 g−1 has been found for HA modified with malonic acid and the lowest surface area of 37 m2 g−1 was calculated for stearic acid modified HA. The BET adsorption average pore size (17–20 nm) of m-HA confirmed its mesoporous nature. The biocompatible nature of the prepared m-HA was assessed by 3-(4,5)-dimethylthiahiazo(-z-yl)-3,5-di-phenytetrazoliumromide (MTT) assay. To evaluate the influence of functional groups and surface properties of m-HA on drug delivery efficiency, ibuprofen was used as a model drug. In vitro drug delivery experimental results indicated that drug loading and release efficiency relied on functional groups, surface area, and porosity of m-HA. The percentage loading of ibuprofen was good for samples containing free –COOH groups and high surface area. A drug loading of 22 mg g−1 has been found for malonic acid modified HA (ma-HA) having high surface area, pore volume, whereas a poor loading of 2.03 mg g−1 has been observed for stearic acid modified HA (st-HA) sample having low surface area and pore volume. A sustained drug release profile showed that 61% drug had been released from malonic acid modified HA (ma-HA) in 24 hours. A 100% drug release was observed for st-HA in 8 hours. Succinic acid modified HA and adipic acid modified HA exhibited intermediate drug release profiles. The drug release behavior of m-HA followed Fick's laws of diffusion.
 Please wait while we load your content...
Please wait while we load your content...

