
Understanding nucleation of the electroactive β-phase of poly(vinylidene fluoride) by nanostructures
Abstract
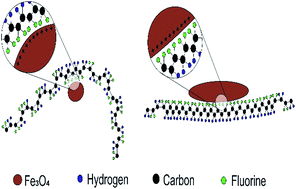
β-Poly(vinylidene fluoride) (PVDF) is of large technological relevance due to its piezoelectric, pyroelectric and/ferroelectric properties. In this way, a variety of methods have been developed to obtain such electroactive β-phase, being the addition of fillers one of the most popular, upscalable and innovative methods. The electrostatic interaction between negative charged fillers with the CH2 groups having a positive charge density has been the most widely accepted mechanism for the direct formation of polar β-phase on nanocomposites. Nevertheless some controversy remains in this matter as the dominating crystallization into the β-phase within PVDF is sometimes attributed to the interaction between the positively charged surfaces of the fillers and the CF2 dipoles in PVDF. In order to clarify such a controversial issue, this work uses two types of nanostructures, Fe3O4 nanorods and Fe3O4 nanoparticles, with distinct sizes and surface charges to study, isolate and evaluate the effects of the different ion–dipole interactions and shapes on the crystalline structures of PVDF. As a result it is shown that in the case of positive ion–CF2 dipole based β-phase nucleation, and beyond the effect of the intermolecular interactions, the rod-shape optimizes the crystallization in the electroactive conformation, thus promoting current development in PVDF-based electroactive devices.
 Please wait while we load your content...
Please wait while we load your content...

