
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The interaction between CuO and Al2O3 and the reactivity of copper aluminates below 1000 °C and their implication on the use of the Cu–Al–O system for oxygen storage and production†
Wenting
Hu‡
 *ab,
Felix
Donat
bc,
S. A.
Scott
a and
J. S.
Dennis
b
*ab,
Felix
Donat
bc,
S. A.
Scott
a and
J. S.
Dennis
b
aDepartment of Engineering, University of Cambridge, Trumpington Street, Cambridge, CB2 1PZ, UK. E-mail: wenting.hu@newcastle.ac.uk
bDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Pembroke Street, Cambridge, CB2 3RA, UK
cLaboratory of Energy Science and Engineering, Department of Mechanical and Process Engineering, ETH Zürich, 8092 Zürich, Switzerland
First published on 22nd November 2016
Abstract
The reversible decomposition of CuO into Cu2O and oxygen at high temperature, typically between 850 and 1000 °C, provides a means of separating pure oxygen from air. In such a process, the oxide generally has to be supported on a refractory oxide, e.g. alumina, to maintain its capacity when cycled many times between CuO and Cu2O. One problem is that if the CuO reacts with the alumina to form CuAl2O4, the latter releases oxygen too slowly to be of practical use so that the capacity for oxygen release of such a carrier falls progressively as more aluminate is formed. However, the reported temperatures at which CuAl2O4 forms are inconsistent so far. This work sets out to investigate the interaction between CuO and different aluminas (and precursors), which are commonly used as support materials, at temperatures between 700 and 1000 °C, as well as some chemical properties of the resulting copper aluminates. It was found that the formation of CuAl2O4 occurred at around 700 °C, 800 °C and 950 °C, when amorphous aluminium hydroxide, γ-alumina, and α-alumina were used as the source of alumina support, respectively. The decomposition of CuAl2O4 in an oxygen-lean environment can lead to the formation of α-alumina as well as γ-alumina, depending on the partial pressure of oxygen. Given that the α-form does not react with CuO around 900 °C, the typical operating temperature for the CuO/Cu2O couple, this observation can be used to partially regenerate CuO from CuAl2O4, for oxygen storage and production at this temperature by decomposing the spinel in a controlled atmosphere to form only α-alumina. However, during the decomposition of CuAl2O4, delafossite CuAlO2 is also formed, limiting the amount of Cu that could be recovered as CuO in a single process cycle.
1. Introduction
CuO is an attractive candidate for the production of oxygen at temperatures around 900 °C owing to its suitable thermodynamic equilibrium as well as the high capacity for the release of gaseous oxygen in| 4CuO(s) ⇌ 2Cu2O(s) + O2(g). | (1) |
The stoichiometric yield of oxygen is about 10 wt% of the CuO. The forward reaction can take place in an oxygen-lean atmosphere, e.g. pure CO2 or steam, and the gaseous oxygen produced (mixed with CO2 or steam) used to burn a solid carbonaceous fuel instead of air. As a result, the dried gaseous products of combustion are largely free of non-condensable gases such as N2 and consist primarily of CO2, which can be subsequently sequestrated with minimal treatment and separation.1,2 The Cu2O produced can be oxidised in air to re-form CuO for repeated use. It is possible to arrange for the combustion of the solid fuel to occur in situ with the oxide releasing oxygen in accordance with the forward reaction of (1) and with the regeneration of CuO taking place in a separate reactor free of fuel. Such a scheme is termed chemical-looping with oxygen uncoupling (CLOU)3 and has been demonstrated in continuous operation with different fuels at various scales.4–6 Alternatively, the combustion can be conducted in a separate combustor, in which case reaction (1) serves as an air separation step. This is known as chemical-looping air separation (CLAS)7 and replaces the energetically-demanding cryogenic separation of oxygen commonly proposed for oxy-fuel combustion.
In practice, pure CuO sinters at temperatures as low as 800 °C (ref. 8) and must be supported on refractory materials with high melting points to retain its reactivity for repeated redox cycles. Al2O3 is a popular choice of refractory because of its abundance and low cost: many copper-based oxygen carriers have been developed with it.9–13 Depending on the method of synthesis, it is possible for CuO and Al2O3 to form the spinel CuAl2O4:
| CuO + Al2O3 → CuAl2O4. | (2) |
The spinel is relatively stable to decomposition. It is therefore important to investigate whether the interaction of CuO and Al2O3 can be inhibited at temperatures around 900 °C to preserve the capacity for oxygen transfer due to reaction (1). The current understanding of the Cu–Al–O system appears incomplete. Firstly, no consistent conclusion has been reached with regard to the lowest temperature at which the formation of CuAl2O4 becomes appreciable. Secondly, and importantly, it is still unclear whether CuAl2O4, once formed, can be decomposed readily into the constituent oxides to restore the capacity for oxygen release. The objective of this paper is to address these two issues, with a particular focus on their relevance to chemical-looping processes for oxygen storage and production.
Amongst previous investigations, Jacob and Alcock14 showed that CuAl2O4 is thermodynamically stable in air at temperatures above ∼600 °C and remains so until at least 1000 °C, depending on whether CuO or Al2O3 is in excess. Misra and Chaklader15 reported the formation of CuAl2O4 from mechanically-mixed fine powders of CuO and abrasive alumina at 800 °C but not at 700 °C. De Diego et al.9 impregnated a porous γ-alumina substrate with a solution of Cu(NO3)2, followed by decomposition of the nitrate at 550 °C for 30 minutes and subsequent calcination in air between 550 °C and 950 °C for 1 hour. The presence of CuAl2O4 was observed in particles calcined at 850 °C or above but not in those calcined at <800 °C. Interestingly, after the particles were subjected at 800 °C to 100 cycles consisting of reduction to metallic Cu with CH4 and re-oxidation with air, CuAl2O4, as well as α-alumina, were detected in all of them regardless of the calcination temperature used to make the particles originally. Imtiaz et al.12 found that a significant amount of CuAl2O4 was formed after co-precipitates of copper and aluminium hydroxides, produced from their respective nitrates and NaOH, were fired at 800 °C for 2 hours. On the other hand, the freeze-granulated particles produced from powders of CuO and α-alumina studied by Arjmand et al.11 only formed a small amount of CuAl2O4 after being calcined at 950 °C for 6 hours, despite having a ratio of Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al much closer to the stoichiometry of CuAl2O4 in the starting mixture. Whilst the spinel can be reduced rapidly to metallic Cu and Al2O3 in the presence of reducing gases such as CH4 and H2 (ref. 9–11 and 16–18) at temperatures around 900 °C, the decomposition of CuAl2O4 in an inert environment is rather slow11,12 and therefore unsuitable for the production of oxygen. Interestingly, the equilibrium partial pressure of O2 (PO2) in
:Al much closer to the stoichiometry of CuAl2O4 in the starting mixture. Whilst the spinel can be reduced rapidly to metallic Cu and Al2O3 in the presence of reducing gases such as CH4 and H2 (ref. 9–11 and 16–18) at temperatures around 900 °C, the decomposition of CuAl2O4 in an inert environment is rather slow11,12 and therefore unsuitable for the production of oxygen. Interestingly, the equilibrium partial pressure of O2 (PO2) in
| 4CuAl2O4(s) ⇌ 4CuAlO2(s) + 2Al2O3(s) + O2(g) | (3) |
| 2CuAl2O4(s) + 2CuO(s) ⇌ 4CuAlO2(s) + O2(g), | (4) |
Of course, one strategy to avoid interaction between CuO and alumina would be to utilise other types of support. For instance, MgAl2O4 (ref. 11, 12 and 19–21) or calcium aluminates, either in the form of commercial cement22 or synthesised from pure materials,23,24 have shown promising performance. However, given that the impregnation of commercial alumina catalyst support is a very convenient method for preparing copper-based carriers, the formation of the spinel merits attention.
2. Experimental
2.1. Preparation of materials
Materials with a nominal composition of 70 wt% CuO and 30 wt% Al2O3 were prepared by mechanical mixing. An appropriate amount of α-alumina powder (Sigma-Aldrich, ≥98%), crushed γ-alumina pellets (originally 3 mm spheres, Alfa Aesar) or powdered Al(OH)3 (Sigma-Aldrich, 50–57% on Al2O3 basis) was mixed with CuO powder (Sigma-Aldrich, ≥98%) in a planetary ball mill (MTI, MSK-SFM-1) operating at 25 Hz for 2 hours to produce a homogeneous mixture. The resulting mixture was calcined in air for 6 hours at a known, constant temperature between 600 °C and 1000 °C for various samples, in a box furnace. For some materials, in particular those containing α-alumina, the milling and calcination process was repeated up to 4 times. These materials are named in a systematic way for easy identification. For instance, a-Al30Cu70-1000-4 denotes the material prepared from α-alumina (g – for γ-alumina and h – for Al(OH)3) and calcined at 1000 °C, 4 times.In addition, CuAl2O4 was synthesised as follows. A 1.0 M solution of NH4HCO3 (Fisher Scientific, 99%) was gradually added to 200 mL of a stirred solution containing 0.10 M Cu(NO3)2 (Sigma-Aldrich, ≥98%) and 0.20 M Al(NO3)3 (Fisher Scientific, ≥98%) until pH 4.0 was reached. The resulting gel was dried at 80 °C overnight and calcined in air at 1000 °C for 6 hours in a box furnace. The solid obtained was subsequently crushed and sieved to <50 μm and 50–100 μm for phase identification and thermogravimetric (TG) analysis, respectively.
2.2. Characterisation of materials
The phases present in the prepared materials were identified from X-ray powder diffraction (XRD; Empyrean PANalytical, Cu-Kα, 40 kV, 40 mA) in the range 2θ = 5–80° with a step size of 0.0167°. The references used for phase identification were: ICSD-31701 (CuAlO2), ICSD-172145 (CuAl2O4), ICSD-52043 (Cu2O), ICSD-16025 (CuO), ICSD-64699 (Cu), ICSD-99836 (tetragonal γ-alumina), ICSD-82504 (θ-alumina) and ICSD-31545 (α-alumina). The reactivity of various materials was characterised using a thermogravimetric analyser (TGA; Mettler Toledo, TGA/DSC1). The TGA furnace was always purged by two streams of Ar (BOC, >99.998%) at a total flowrate of 100 mL min−1 in addition to a stream of reactive gas with a flowrate of ∼50 mL min−1. The flowrates are as measured at 20 °C and atmospheric pressure, and this applies to all flowrates quoted in the following text, unless specified otherwise. The reactive gas could be switched between air (BOC, >99.995%), N2 (BOC, >99.998%) or 5 vol% H2 balanced by N2 (BOC, >99.999%; referred to simply as H2 hereafter) using a solenoid valve manifold (Bürkert, type 6011) controlled from a separate program synchronised with the TGA.Two types of TG experiments were undertaken on fresh materials. In the first, to quantify the total amount of Cu (on a CuO basis) and CuAl2O4 present in the prepared materials, approximately 20 mg of powdered sample was subjected to isothermal reaction at 900 °C with a reactive gas sequence air–N2–H2–air, each lasting 15 minutes. In the second, to investigate the reactivity of pure CuAl2O4, approximately 20 mg of the synthesised samples were held at 1000 °C, initially in air. Once the temperature and the mass of the sample had stabilised, the atmosphere was switched to a mixture of air and N2, resulting in a partial pressure of O2 ranging from 0–0.02 bar for 50 minutes. It should be noted that when no air was used, the partial pressure of O2 in the TGA furnace was <5 × 10−4 bar. The exact partial pressure of O2 was determined by measuring the equilibrium temperature of the reversible reaction (1) under the same atmosphere as that used in each experiment with CuAl2O4. The stabilised CuO-based particles have been characterised in previous work.24 Some samples of CuAl2O4 with higher masses, ∼60 mg each, were treated using a combination of the above protocols (with some variations in duration or gas composition, when necessary) to investigate the redox behaviour of the material further. Identification of the solid phases present in some of the recovered samples was also performed using XRD. The details of the changes made to the TG protocols will be specified along with the results in the next section.
3. Results
3.1. The effect of different alumina precursors on the formation of CuAl2O4
The formation of CuAl2O4 was found to occur at different temperatures, depending on the form of alumina (or precursor) used. α-Alumina did not react appreciably with CuO at 900 °C, even after four repetitions of milling and calcination for 6 hours on the same sample, as confirmed by the absence of peaks belonging to CuAl2O4 in the XRD diffractograms shown in Fig. 1. However, when calcined in air at the higher temperature of 1000 °C for 6 hours, a significant amount of CuAl2O4 was formed and further milling and calcining led to the formation of CuAlO2 with almost all the free alumina consumed by the 4th calcination. When calcined at 950 °C, a small amount of CuAl2O4 but no CuAlO2 was detected in the material. This observation is in agreement with the finding of Jacob and Alcock that in an atmosphere with PO2 = 0.21 bar, reaction (4) has an equilibrium temperature of 1003 °C.14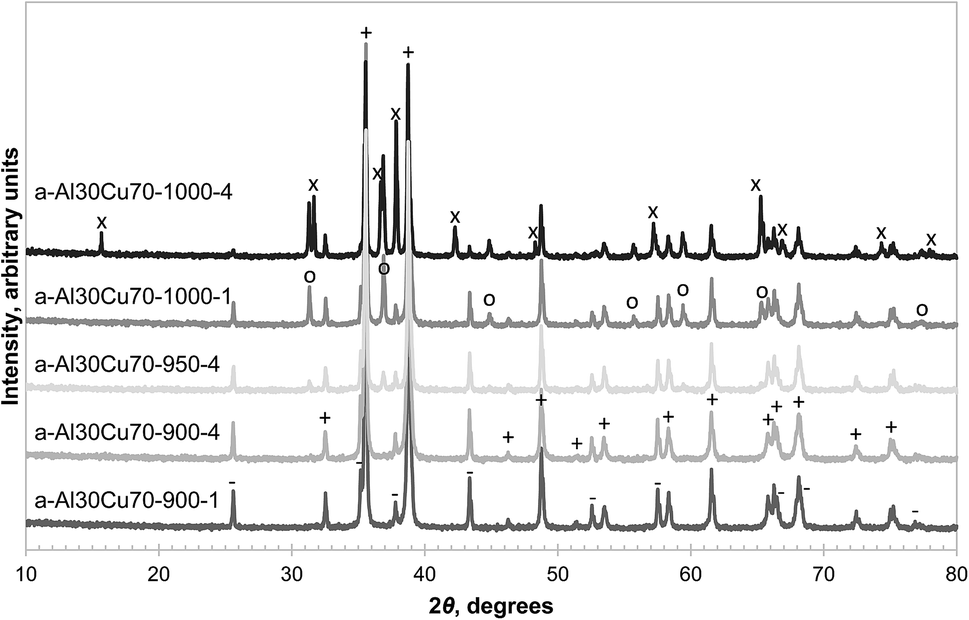 | ||
| Fig. 1 XRD diffractograms of selected fresh samples prepared from α-alumina with the major reflection peaks annotated as: CuO (+), α-Al2O3 (−), CuAl2O4 (○), CuAlO2 (×). | ||
In contrast, Fig. 2 shows that, after heating in air at 850 °C for 6 hours, the formation of CuAl2O4 from γ-alumina and CuO was significant. Since γ-alumina is only weakly reflecting and therefore difficult to quantify by XRD, the conversion of alumina to CuAl2O4 was determined by measuring the changes in mass of a sample in a TGA at 900 °C, exposed successively to air, N2, H2 and finally air. As an example, the result for g-Al30Cu70-900-1 is shown in Fig. 3. It can be seen that the CLOU capacity of g-Al30Cu70-900-1 was ∼5.3 wt% and the total oxygen capacity of the material was 14.5 wt%, corresponding to a Cu content of 72.5 wt% on a CuO basis, instead of the expected value of 70 wt%. The difference was probably owing to adsorbed moisture in the γ-alumina used during the initial preparation. During the decomposition under N2, the material exhibited a fast reaction stage between 900 and 1300 s, followed by a very slow stage until the atmosphere was switched to H2. As will be discussed in Section 3.2., the reduction of CuAl2O4 to CuAlO2 in an inert environment via reaction (3) is extremely slow. Thus it is plausible that the first stage is dominated by reaction (1) and the second stage by the slow reaction (3). In fact, the chemical kinetics of decomposition of CuO in N2 is faster than that being measured in this experiment24 and here the rate is primarily limited by mass transfer in the TGA. A more detailed discussion is presented in the ESI.† The amount of free CuO present in g-Al30Cu70-900-1, which amounts to 52.8 wt%, can be estimated from the mass change during the fast stage. Assuming all remaining Cu is bound in the form of CuAl2O4, 91.8% of the γ-alumina present initially would have had to have been consumed during the calcination when the material was first prepared. The same analysis carried out on a sample of a-Al30Cu70-950-4 showed that only 2.4% of the α-alumina had reacted during its preparation, albeit the material having been calcined at a higher temperature of 950 °C for a period 4 times longer.
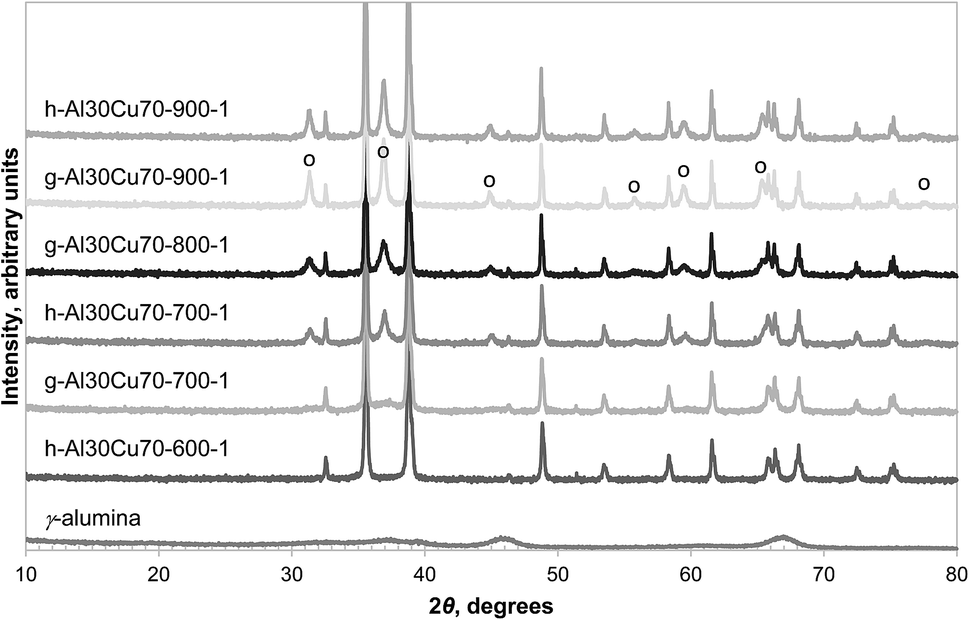 | ||
| Fig. 2 XRD diffractograms of selected fresh samples prepared from γ-alumina and Al(OH)3. The reflection peaks of CuAl2O4 are annotated (○) and the remaining peaks are due to CuO, with the exception of the γ-alumina sample at the bottom of the figure. | ||
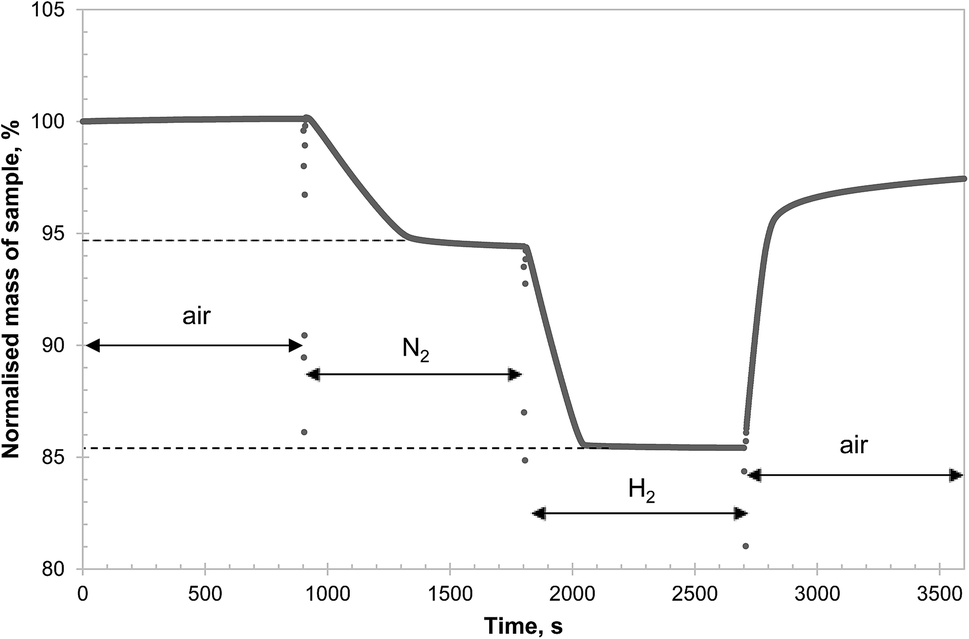 | ||
| Fig. 3 TG curve of the isothermal reaction of g-Al30Cu70-900-1 in different atmospheres at 900 °C. The abrupt changes in mass seen between different segments were due to gas switching, disturbing the microbalance. | ||
Amongst the three precursors investigated, Al(OH)3 was found to be the most reactive, capable of forming CuAl2O4 at temperatures as low as 700 °C, as seen in Fig. 2. It should be noted that the Al(OH)3 used in this work was amorphous, as confirmed by XRD (shown in Fig. S1 in the ESI†), as was the sample dehydrated at 600 °C for 24 hours. A small amount of γ-alumina was formed after Al(OH)3 was calcined in air at 700 °C for 6 hours. Further characterisation of the Al(OH)3 sample was undertaken using temperature programmed decomposition (TPD) in air in the TGA, with a heating rate of 2 °C min−1. The result is shown in Fig. 4. It can be seen that the hydroxide gradually lost H2O until ∼600 °C and a marked decomposition occurred around 800 °C. It has been reported that amorphous alumina transforms to γ-alumina at 802 °C and subsequently to α-alumina at 1087 °C.25 In fact, when a sample of Al(OH)3 was calcined at 900 °C in air for 6 hours, the resulting material was confirmed as γ-alumina (as shown in Fig. S1 in the ESI†). These results suggest that amorphous alumina is able to retain some OH groups up to 800 °C, which are lost when the phase transition to γ-alumina occurs.
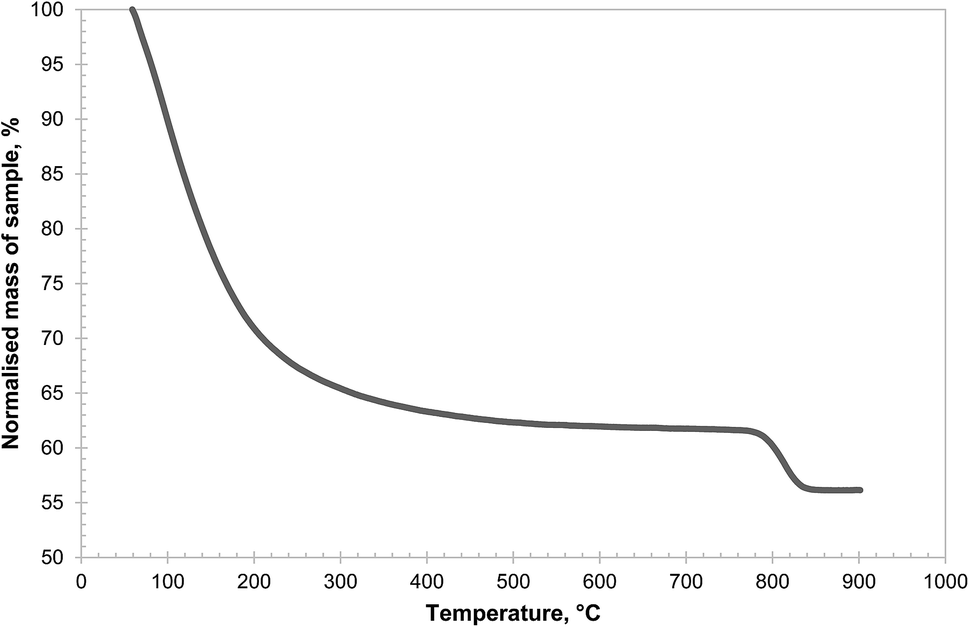 | ||
| Fig. 4 Temperature programmed decomposition of Al(OH)3 sample in air, using the TGA. A constant heating rate of 2 °C min−1 was used and the mass of solid remaining at 900 °C was 56.1 wt%, in line with the material specification. | ||
3.2. The thermal decomposition of CuAl2O4
CuAl2O4 was synthesised from the decomposition of nitrate precursors because (i) aluminium nitrate forms amorphous alumina on heating26 and (ii) the gel method used ensured that the Cu and Al precursors were homogeneously mixed before firing at 1000 °C. XRD analysis, given in Fig. 5, confirmed that the synthesised sample was comprised primarily of CuAl2O4 with trace amounts of CuO and α-alumina. The decomposition of CuAl2O4 in Ar at 900 °C, shown in Fig. S2 of the ESI,† was found to be slow and was not complete even after 20 hours. On the other hand, a similar degree of conversion was achieved in 50 minutes when the material was decomposed at 1000 °C, and the results are given in Fig. S3 of the ESI.† Based on these limited data of the isothermal decomposition, it appears that a high activation energy is associated with the reaction. Assuming the decomposition is due to reaction (3) alone, an apparent activation energy of the reaction can be estimated knowing the rate of change of conversion of the solid at various iso-conversion points (the procedure is briefly described in the ESI†). These values are plotted in Fig. S4 of the ESI,† up to a conversion of 0.8. The values were found to be fairly constant at ∼370 kJ mol−1 between conversions of 0.25 and 0.75. At lower values of conversion, the apparent activation energy was much lower. It may be possible that in the early stage of the decomposition, the rate-determining step is different from that at a later stage but this cannot be ascertained with the current data and further investigation is beyond the scope of this paper. It is worth noting that for a gas–solid reaction like reaction (3), the enthalpy of reaction is a measure of how fast the thermodynamic driving force changes with temperature in an inert gas atmosphere. Here, the apparent activation energy was found to be much higher than the enthalpy of reaction, ∼140 kJ mol−1,14 suggesting that chemical kinetics has a larger effect than thermodynamics on the observed rate of reaction with changing temperature.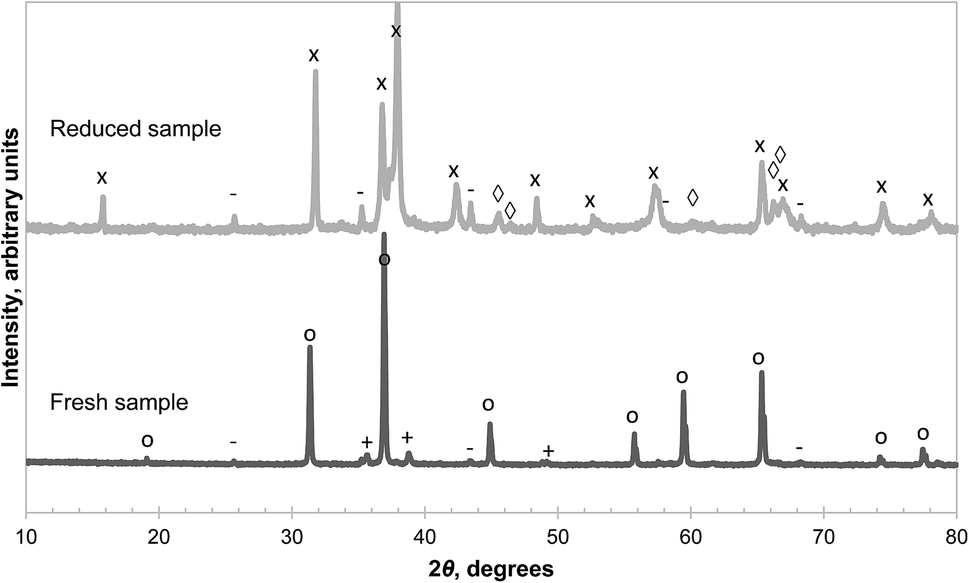 | ||
| Fig. 5 XRD diffractograms of synthesised CuAl2O4 as prepared (bottom) and after being decomposed in Ar at 900 °C for 20 hours (top). The major reflection peaks are annotated as: CuO (+), α-Al2O3 (−), CuAl2O4 (○), CuAlO2 (×), γ-Al2O3 (◊). | ||
Examination of the reduced sample (recovered after decomposition at 900 °C and cooled in Ar) revealed CuAlO2, α-alumina and γ-alumina as the main phases present as seen in Fig. 5. Cu2O was not detected in the reduced sample, contrary to the findings of Arjmand et al.,18 who used CH4 instead of an inert gas. Interestingly, the reflections from γ-alumina are more consistent with a tetragonal structure, evident from the splitting of the peak around 2θ = 46°, instead of the commonly-adopted cubic structure.27 The phase obtained from the thermal decomposition of the synthetic CuAl2O4 also appears to be more crystalline than the as-received γ-alumina, or that derived from amorphous alumina, the XRD diffractograms of which, given respectively in Fig. 2, and in Fig. S1 of the ESI,† contain much broader peaks. The amounts of various species present in the reduced sample were estimated by quantitative analysis of the XRD diffractogram and the results are shown in Table 1. The results are in good agreement with an independent estimate made from the thermogravimetric analysis, assuming that the unreacted sample consisted of pure CuAl2O4 and the mass loss was due to the evolution of gaseous O2 according to reaction (3). The fact that a significant amount of γ-alumina was formed from the decomposition of CuAl2O4 raises questions about the thermodynamics of reaction (3) because its standard Gibbs free energy of reaction at 900 °C is reported as 41.5 ± 1.3 kJ mol−1 (ref. 14) whereas the Gibbs free energy of transformation from γ-alumina to α-alumina is approximately −14 kJ mol−1 at the same temperature.28 Consequently, the equilibrium PO2 of reaction (3) would be rather different depending on the polymorph of the product alumina.
| Species | Percentage by weight | ||
|---|---|---|---|
| XRD analysis | Thermogravimetry | ||
| CuAl2O4 | 12.8% | 12.2% | |
| CuAlO2 | 65.5% | 62.0% | |
| γ-Alumina | 7.7% | 21.7% | 25.8% |
| α-Alumina | 14.0% | ||
The results in Fig. 5 can be explained by two possible reaction schemes. In the first, CuAl2O4 decomposes to both polymorphs of alumina in parallel, each having a different equilibrium PO2; in the second, CuAl2O4 decomposes to γ-alumina first, which then transforms to α-alumina. Further investigations were carried out to distinguish between the two schemes and the results are presented in Fig. 6. From the figure, it can be seen that the initial mass loss (over 160 s) of CuAl2O4 when decomposed at 1000 °C in PO2 up to 0.02 bar showed two distinct linear regimes. The change of the average rate of mass loss with respect to PO2 was significantly faster when PO2 < 0.009 bar. This result supports the hypothesis that the decomposition of CuAl2O4 to the two forms of alumina occurs in parallel. Had the reaction followed the sequence CuAl2O4 → γ-alumina → α-alumina, one would not expect such sudden change in the rate of decomposition with respect to PO2. As the decomposition proceeded rather slowly, external mass transfer did not have a significant influence on the measured rate of reaction: the estimated difference of PO2 between the surface of the samples and that above the crucible holding the sample (4.9 mm diameter and 4 mm deep) using the fastest rate measured (∼0.135 mg over 160 s, as shown in Fig. 6) was approximately 1.5 × 10−5 bar. Thus, assuming that the rate of decomposition of CuAl2O4 was proportional to the difference between PO2 of the gas environment and the equilibrium value, the equilibrium PO2 of the two parallel reactions can be estimated from the intersection between the two regimes, 8.7 × 10−3 bar, and that between the slow regime and the abscissa, 0.034 bar. The difference in the equilibrium PO2 amounts to a difference in the Gibbs free energy of 14.4 kJ mol−1 of reaction (3), assuming the activities of the solids are unity. However, this value is only ∼60% of the Gibbs free energy of transformation from γ-alumina to α-alumina reported in the literature.28 The discrepancy could be due to the tetragonal deformation of the γ-alumina having a lower Gibbs free energy of formation. In fact, at the temperature concerned, it is possible for γ-alumina to start transforming to other transition aluminas,29 which is probably why a tetragonal deformation, rather than the more common cubic structure, was observed from the decomposition of CuAl2O4. On a side note, using the value of 0.034 bar, the Gibbs free energy of reaction (3) with α-alumina as the product is estimated as 35.6 kJ mol−1, slightly higher than the value of 33.0 kJ mol−1, given by Jacob and Alcock,14 although the corresponding difference in PO2 is more than 20%.
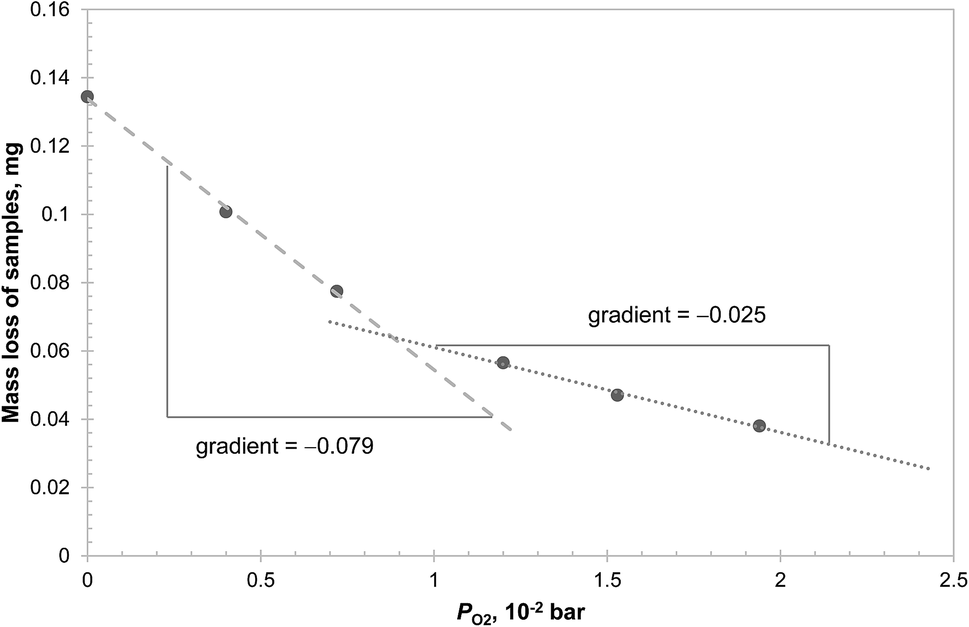 | ||
| Fig. 6 Loss of mass of samples of CuAl2O4 during the initial 160 s of isothermal decomposition at 1000 °C in different PO2. The mass of sample used in each experiment was between 19.46 mg and 19.67 mg. | ||
In addition, two samples of CuAl2O4 were decomposed at 1000 °C in the TGA for 3 hours, one with PO2 = 9.0 × 10−3 bar, just above the transition point seen in Fig. 6, and the other in a mixture of N2 and Ar (where PO2 < 5 × 10−4 bar due to a small leakage of air into the TGA). The XRD diffractograms of the decomposed samples, given in Fig. 7, revealed that in the former case, some CuAl2O4 remained but no γ-alumina was detected, whereas the converse is true for the latter. These observations also suggest that the decomposition of CuAl2O4 to different phases of alumina probably occurs in parallel rather than in series.
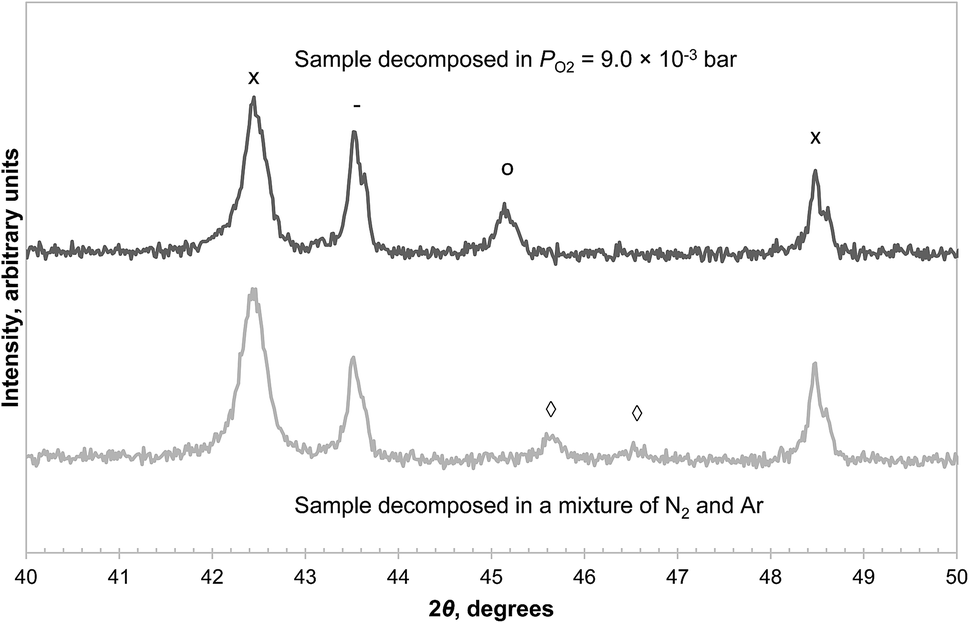 | ||
| Fig. 7 XRD diffractograms of CuAl2O4 decomposed at 1000 °C in different atmospheres. Only a section between 2θ = 40° and 50° is shown to distinguish the peak due to CuAl2O3 at 45.2° and that due to γ-Al2O3 at 45.6°. The reflection peaks are annotated as α-Al2O3 (−), CuAl2O4 (○), CuAlO2 (×), γ-Al2O3 (◊). A separate graph containing the diffractograms between 2θ = 10° and 80° can be found in the ESI (Fig. S5†). | ||
3.3. The oxidation and reduction of CuAlO2
There are two potential pathways for the oxidation of CuAlO2, i.e. the backward reactions of (3) and (4). The equilibrium PO2 of reaction (4) is significantly higher than that of reaction (3) in the temperature range of interest, between 900 °C and 1000 °C.14 Therefore the two pathways can be examined separately by choosing an appropriate PO2 so that only the backward reaction of (3) is thermodynamically feasible. Here, a sample of CuAl2O4 was decomposed in a mixture of N2 and Ar at 1000 °C for 50 minutes, followed by oxidation in an atmosphere with PO2 ≈ 0.10 bar (the equilibrium PO2 of the reverse reaction (3) is ∼0.04 bar with α-alumina as the reactant and that of reaction (4) is above 0.9 bar (ref. 14)) at the same temperature for a further 50 minutes and the result is shown in Fig. S6 of the ESI.† It was found that the oxidation of CuAlO2via reaction (3) was much slower than the forward decomposition reaction, despite having a slightly higher thermodynamic driving force. The kinetics of the backward reaction (4) were not investigated in this work, as Arjmand et al. have shown previously that complete oxidation of CuAlO2via this route was only achieved after prolonged heating in air.18 It appears that both routes for the oxidation of CuAlO2 are slow and the material does not contribute to the oxygen carrying capacity significantly, bearing in mind that many chemical-looping processes oxidise the oxygen carrier in a circulating fluidised bed, which normally has a low residence time.On the other hand, the reduction of CuAlO2 in H2 was fast. For instance, a sample of CuAl2O4 first decomposed in an atmosphere with PO2 = 9.0 × 10−3 bar for 10 hours at 1000 °C was reduced completely in 5.5 minutes once the reactive gas was switched to 5% H2. The average rate of loss of mass during this time was approximately 0.6 mg min−1. This is equivalent to 38 μmol min−1 of oxygen atoms, and is comparable to the rate of transfer of H2 to the solid estimated in previous work for the same experimental arrangement, i.e. the reaction was limited by mass transfer.23 XRD analysis of the reduced sample showed that the phases present were metallic Cu, θ-alumina and α-alumina, as annotated in Fig. 8. Several studies30–32 suggest that the reduction of CuAlO2 at around 1000 °C results in interspersed Cu and θ-alumina without any α-alumina being formed. The α-alumina present in this work is probably the result of the decomposition of CuAl2O4 only and this is supported by a subsequent TG experiment, which is shown in Fig. 9 and will be discussed later. However, no attempt was made to synthesise pure CuAlO2 to confirm this, it not being the primary focus of the current work.
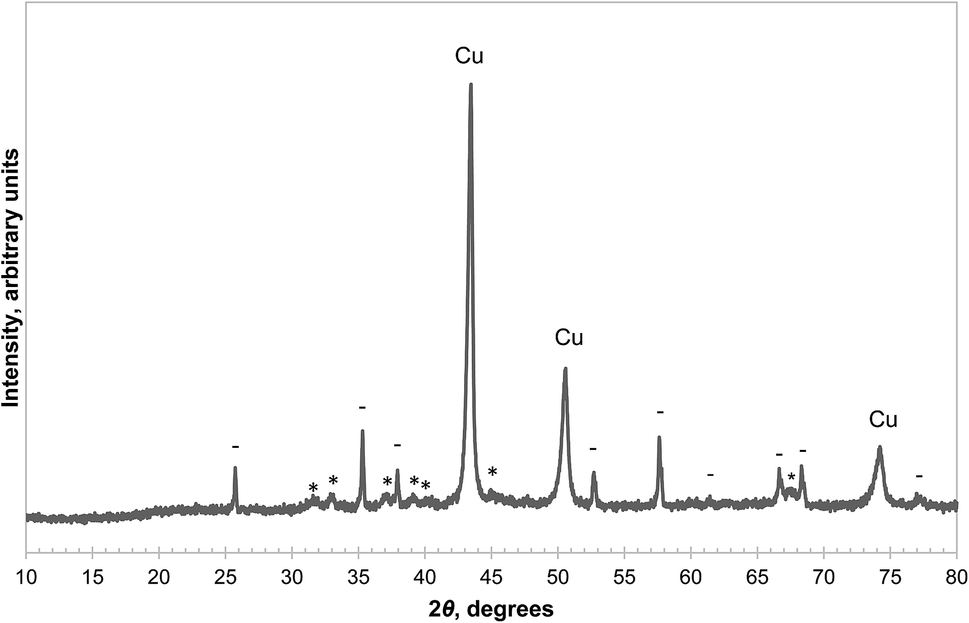 | ||
| Fig. 8 XRD diffractograms of a sample of CuAl2O4 first decomposed in 0.90% O2 at 1000 °C for 10 hours, then reduced by H2. The annotations are: metallic Cu (Cu), α-Al2O3 (−), θ-alumina (*). | ||
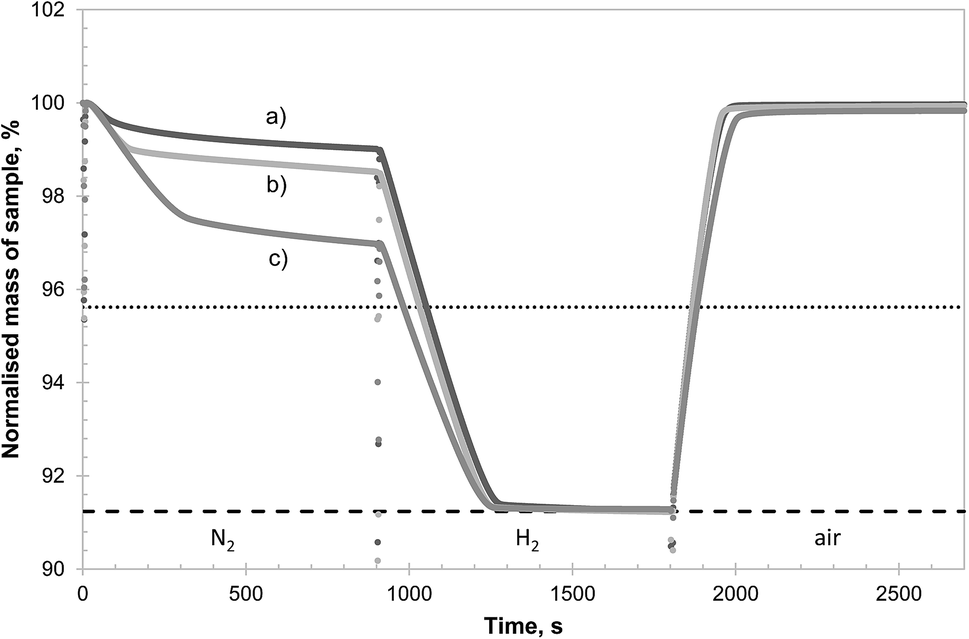 | ||
| Fig. 9 TG curve of the isothermal reaction of 3 different samples of CuAl2O4 in different atmospheres at 900 °C (N2, H2 and air). The samples were kept in air before the start of the experiment to maintain a fully oxidised state. The abrupt changes in mass seen between different segments were due to gas switching, disturbing the microbalance. The dotted and dashed horizontal lines indicate, respectively, the theoretical mass change if all the Cu2+ present is reduced to Cu+ and metallic Cu. The 3 samples were: (a) CuAl2O4 as synthesised (60.89 mg), (b) CuAl2O4 fully reduced in H2 and re-oxidised in air at 900 °C prior to the experiment (61.37 mg) and (c) CuAl2O4 first decomposed in 0.90% O2 at 1000 °C for 10 hours, then reduced by H2 (i.e. the same as in Fig. 8) followed by re-oxidation at 900 °C prior to experiment (61.11 mg). | ||
4. Discussion
The results presented in this work concur with previous findings that the generation of gas phase oxygen by the decomposition of the spinel phase, CuAl2O4, is slow up to 1000 °C (ref. 11 and 12) and therefore unsuitable for oxygen production schemes such as CLOU or CLAS. Thus, the formation of the spinel must be prevented to retain the high reactivity of CuO with regard to oxygen release. Since the typical operating temperature for the CuO/Cu2O system is around 900 °C, it is viable to use α-alumina as a support material but not amorphous alumina or γ-alumina, which react with CuO at much lower temperatures. Usually the latter two are preferred as support materials since they possess much higher internal surface areas compared with α-alumina. However, because the operating temperature is sufficiently high, a high surface area is not necessary to secure a high reaction rate and this has been demonstrated in previous work for various metal oxide oxygen carriers.6,23,33 On the other hand, when high surface area is desired, e.g. for catalysis at intermediate or low temperatures, amorphous alumina and γ-alumina can be used as long as the operating temperature is below 600 °C and 700 °C, respectively, where the formation of CuAl2O4 from CuO and respective alumina does not occur, as seen in Fig. 2. In any case, alumina supports should be avoided when the operating temperature is constantly above 950 °C since even the relatively-stable α-alumina starts to form CuAl2O4 at this temperature.The fact that CuAl2O4 decomposes to different polymorphs of alumina depending on the PO2 of the environment can be exploited to regenerate the active CuO phase should the spinel be accidentally formed. The scheme is proposed as follows: first, the material containing the spinel phase would be maintained in an atmosphere with appropriate PO2 (e.g. slightly above 9.0 × 10−3 bar if treated at 1000 °C, the transition point shown in Fig. 6) to slowly decompose the spinel into α-alumina and the delafossite phase, CuAlO2, while avoiding the formation of γ-alumina. Once the decomposition is complete, the material would be further reduced by a fuel gas (e.g. CH4 or syngas) at 900 °C or below, to minimise the sintering of the metallic Cu formed (the melting point of Cu is lower than its oxides, or the aluminates, at 1084 °C). The reduced material would then be re-oxidised to regenerate the CuO phase, preferably at low temperature and PO2, e.g. ∼700 °C and 0.01 bar, because particles containing Cu agglomerate easily when being oxidised in air at high temperatures.9
To demonstrate the effectiveness of this procedure, a comparative study of CuAl2O4 with different pre-treatments was performed in the TGA and the results are shown in Fig. 9. It can be seen from Fig. 9 that as-synthesised CuAl2O4 (sample a) released a limited amount of oxygen in the first 15 minutes of the experiment and reducing the sample in H2 and re-oxidised once (sample b) did not bring significant improvement. However, the sample decomposed to CuAlO2 before being reduced in H2 (sample c) was able to release much more oxygen during the inert stage. All three samples were capable of being reduced completely in H2 and re-oxidised fully in air within minutes. The rates of reduction and oxidation were again limited by external mass transfer and did not reflect the reactivity of the materials. The slightly slower rates of reduction and oxidation observed for (sample c) were due to a slight increase in the flow rate of the purging Ar during this particular experiment, resulting in a higher dilution of the H2, rather than the deactivation of the material. This was confirmed by identical rates of decomposition of the CuO observed for all materials during the decomposition in N2, which is not affected by dilution of gases. Using the same analysis as undertaken in Fig. 3, it was estimated that approximately 6% of the Cu was present as CuO in sample (a) and the corresponding percentages in samples (b) and (c) were 23% and 55%, respectively. Assuming the additional CuO present in sample (c) arose entirely from the formation of α-alumina (which does not reform CuAl2O4 on subsequent oxidation), and that the decomposition of CuAl2O4 to CuAlO2 produces α-alumina exclusively, the expected amount of CuO in sample (c) would be 53% of the total Cu, very close to the value measured experimentally. In conjunction with results shown in Fig. 8, it can be inferred that the alumina formed from the reduction of CuAlO2 should be mostly, if not all, composed of the θ-phase. Furthermore, θ-alumina was able to react with the interspersed Cu to form CuAl2O4 directly on oxidation without forming CuAlO2 as an intermediate—otherwise the oxidation would not have been complete. Owing to the presence of θ-alumina, it would not be possible to recover all the Cu as CuO following the proposed protocol but 50% of the Cu bound in CuAl2O4 could be recovered and if repeated several times, most of the Cu might be re-activated into the CuO phase.
The series of interactions between CuO and alumina investigated in this work is summarised in Fig. 10.
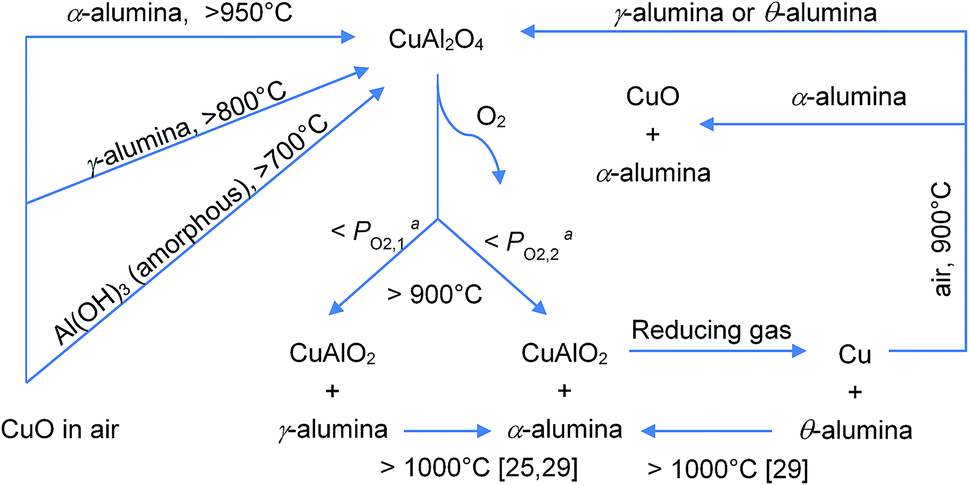 | ||
| Fig. 10 Summary of possible reactions involving alumina and CuO and related compounds. aPO2,1 < PO2,2. | ||
5. Conclusion
The formation of CuAl2O4 from CuO and alumina was investigated, together with its decomposition in an oxygen-lean environment. The oxidation and reduction characteristics of the related copper aluminate, CuAlO2, was also studied. It was found that(1) The solid–solid reaction between CuO and different polymorphs of alumina occurs at different temperatures. The ease of reaction follows the order amorphous alumina > γ-alumina > α-alumina.
(2) It is possible to support CuO with α-alumina for oxygen storage and production up to 950 °C without forming the undesired CuAl2O4 phase, whereas aluminium hydroxide (and its derivative amorphous alumina), γ-alumina and θ-alumina are unsuitable for this purpose.
(3) CuAl2O4 can decompose slowly to form both α-alumina and γ-alumina. By controlling the PO2 of the environment, it is possible to bias the selectivity towards 100% α-alumina so that on subsequent oxidation of the material, CuO could be formed instead of CuAl2O4.
(4) CuAlO2 resists oxidation in air but can be easily reduced in H2. θ-Alumina is formed in the reduction process.
(5) Based on the reactivity of CuAl2O4 and CuAlO2, a possible procedure for the regeneration of CuO from copper aluminates is proposed, which could recover up to 50% of the copper originally bound in the form of CuAl2O4 in a single treatment.
Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC Grants EP/I010912/1 and EP/K030132/1).References
- B. J. P. Buhre, L. K. Elliott, C. D. Sheng, R. P. Gupta and T. F. Wall, Oxy-fuel combustion technology for coal-fired power generation, Prog. Energy Combust. Sci., 2005, 31, 283–307, DOI:10.1016/j.pecs.2005.07.001.
- M. B. Toftegaard, J. Brix, P. A. Jensen, P. Glarborg and A. D. Jensen, Oxy-fuel combustion of solid fuels, Prog. Energy Combust. Sci., 2010, 36, 581–625, DOI:10.1016/j.pecs.2010.02.001.
- T. Mattisson, A. Lyngfelt and H. Leion, Chemical-looping with oxygen uncoupling for combustion of solid fuels, Int. J. Greenhouse Gas Control, 2009, 3, 11–19, DOI:10.1016/j.ijggc.2008.06.002.
- A. Abad, I. Adánez-Rubio, P. Gayán, F. García-Labiano, L. F. de Diego and J. Adánez, Demonstration of chemical-looping with oxygen uncoupling (CLOU) process in a 1.5 kWth continuously operating unit using a Cu-based oxygen-carrier, Int. J. Greenhouse Gas Control, 2012, 6, 189–200, DOI:10.1016/j.ijggc.2011.10.016.
- I. Adánez-Rubio, A. Abad, P. Gayán, L. F. de Diego, F. García-Labiano and J. Adánez, Biomass combustion with CO2 capture by chemical looping with oxygen uncoupling (CLOU), Fuel Process. Technol., 2014, 124, 104–114, DOI:10.1016/j.fuproc.2014.02.019.
- I. Adánez-Rubio, A. Abad, P. Gayán, L. F. de Diego, F. García-Labiano and J. Adánez, Performance of CLOU process in the combustion of different types of coal with CO2 capture, Int. J. Greenhouse Gas Control, 2013, 12, 430–440, DOI:10.1016/j.ijggc.2012.11.025.
- B. Moghtaderi, Application of Chemical Looping Concept for Air Separation at High Temperatures, Energy Fuels, 2010, 24, 190–198, DOI:10.1021/ef900553j.
- L. F. de Diego, F. García-Labiano, J. Adánez, P. Gayán, A. Abad, B. M. Corbella and J. María Palacios, Development of Cu-based oxygen carriers for chemical-looping combustion, Fuel, 2004, 83, 1749–1757, DOI:10.1016/j.fuel.2004.03.003.
- L. F. de Diego, P. Gayán, F. García-Labiano, J. Celaya, A. Abad and J. Adánez, Impregnated CuO/Al2O3 Oxygen Carriers for Chemical-Looping Combustion: Avoiding Fluidized Bed Agglomeration, Energy Fuels, 2005, 19, 1850–1856, DOI:10.1021/ef050052f.
- S. Chuang, J. Dennis, A. Hayhurst and S. Scott, Development and performance of Cu-based oxygen carriers for chemical-looping combustion, Combust. Flame, 2008, 154, 109–121, DOI:10.1016/j.combustflame.2007.10.005.
- M. Arjmand, A.-M. Azad, H. Leion, A. Lyngfelt and T. Mattisson, Prospects of Al2O3 and MgAl2O4-Supported CuO Oxygen Carriers in Chemical-Looping Combustion (CLC) and Chemical-Looping with Oxygen Uncoupling (CLOU), Energy Fuels, 2011, 25, 5493–5502, DOI:10.1021/ef201329x.
- Q. Imtiaz, M. Broda and C. R. Müller, Structure–property relationship of co-precipitated Cu-rich, Al2O3- or MgAl2O4-stabilized oxygen carriers for chemical looping with oxygen uncoupling (CLOU), Appl. Energy, 2014, 119, 557–565, DOI:10.1016/j.apenergy.2014.01.007.
- Q. Song, W. Liu, C. D. Bohn, R. N. Harper, E. Sivaniah, S. A. Scott and J. S. Dennis, A high performance oxygen storage material for chemical looping processes with CO2 capture, Energy Environ. Sci., 2013, 6, 288–298, 10.1039/c2ee22801g.
- K. T. Jacob and C. B. Alcock, Thermodynamics of CuAlO2 and CuAl2O4 and Phase Equilibria in the System Cu2O–CuO–Al2O3, J. Am. Ceram. Soc., 1975, 58, 192–195, DOI:10.1111/j.1151-2916.1975.tb11441.x.
- S. K. Misra and A. C. D. Chaklader, The System Copper Oxide–Alumina, J. Am. Ceram. Soc., 1963, 46, 509, DOI:10.1111/j.1151-2916.1963.tb13788.x.
- P. Cho, T. Mattisson and A. Lyngfelt, Comparison of iron-, nickel-, copper- and manganese-based oxygen carriers for chemical-looping combustion, Fuel, 2004, 83, 1215–1225, DOI:10.1016/j.fuel.2003.11.013.
- Q. Imtiaz, A. M. Kierzkowska and C. R. Müller, Coprecipitated, copper-based, alumina-stabilized materials for carbon dioxide capture by chemical looping combustion, ChemSusChem, 2012, 5, 1610–1618, DOI:10.1002/cssc.201100694.
- M. Arjmand, A.-M. Azad, H. Leion, T. Mattisson and A. Lyngfelt, Evaluation of CuAl2O4 as an Oxygen Carrier in Chemical-Looping Combustion, Ind. Eng. Chem. Res., 2012, 51, 13924–13934, DOI:10.1021/ie300427w.
- I. Adánez-Rubio, P. Gayán, A. Abad, L. F. de Diego, F. García-Labiano and J. Adánez, Evaluation of a Spray-Dried CuO/MgAl2O4 Oxygen Carrier for the Chemical Looping with Oxygen Uncoupling Process, Energy Fuels, 2012, 26, 3069–3081, DOI:10.1021/ef3002229.
- M. Arjmand, M. Keller, H. Leion, T. Mattisson and A. Lyngfelt, Oxygen Release and Oxidation Rates of MgAl2O4-Supported CuO Oxygen Carrier for Chemical-Looping Combustion with Oxygen Uncoupling (CLOU), Energy Fuels, 2012, 26, 6528–6539, DOI:10.1021/ef3010064.
- P. Gayán, I. Adánez-Rubio, A. Abad, L. F. de Diego, F. García-Labiano and J. Adánez, Development of Cu-based oxygen carriers for Chemical-Looping with Oxygen Uncoupling (CLOU) process, Fuel, 2012, 96, 226–238, DOI:10.1016/j.fuel.2012.01.021.
- L. Xu, J. Wang, Z. Li and N. Cai, Experimental Study of Cement-Supported CuO Oxygen Carriers in Chemical Looping with Oxygen Uncoupling (CLOU), Energy Fuels, 2013, 27, 1522–1530, DOI:10.1021/ef301969k.
- F. Donat, W. Hu, S. A. Scott and J. S. Dennis, Characteristics of Copper-based Oxygen Carriers Supported on Calcium Aluminates for Chemical-Looping Combustion with Oxygen Uncoupling (CLOU), Ind. Eng. Chem. Res., 2015, 54, 6713–6723, DOI:10.1021/acs.iecr.5b01172.
- W. Hu, F. Donat, S. A. Scott and J. S. Dennis, Kinetics of oxygen uncoupling of a copper based oxygen carrier, Appl. Energy, 2016, 161, 92–100, DOI:10.1016/j.apenergy.2015.10.006.
- F. Abbattista, S. Delmastro, G. Gozzelino, D. Mazza, M. Vallino, G. Busca, V. Lorenzelli and G. Ramis, Surface characterization of amorphous alumina and its crystallization products, J. Catal., 1989, 117, 42–51, DOI:10.1016/0021-9517(89)90219-4.
- B. Pacewska and M. Keshr, Thermal transformations of aluminium nitrate hydrate, Thermochim. Acta, 2002, 385, 73–80, DOI:10.1016/s0040-6031(01)00703-1.
- G. Paglia, C. E. Buckley, A. L. Rohl, B. A. Hunter, R. D. Hart, J. V. Hanna and L. T. Byrne, Tetragonal structure model for boehmite-derived γ-alumina, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 144110, DOI:10.1103/physrevb.68.144110.
- M. Chase, Nist-Janaf thermochemical tables, American Institute of Physics, Woodbury, New York, 1998 Search PubMed.
- K. Wefers and C. Misra, Oxides and Hydroxides of Aluminum, Alcoa Tech. Pap., 19, 1987, pp. 1–100, http://www.alcoa.com/global/en/innovation/papers_patents/pdf/TP19_Wefers.pdf, accessed 12 July, 2016 Search PubMed.
- M. Kracum, A. Kundu, M. P. Harmer and H. M. Chan, Novel interpenetrating Cu–Al2O3 structures by controlled reduction of bulk CuAlO2, J. Mater. Sci., 2014, 50, 1818–1824, DOI:10.1007/s10853-014-8744-8.
- D. Byrne, A. Cowley, P. McNally and E. McGlynn, Delafossite CuAlO2 film growth and conversion to Cu–Al2O3 metal ceramic composite via control of annealing atmospheres, CrystEngComm, 2013, 15, 6144, 10.1039/c3ce40197a.
- Z. Yu, M. Kracum, A. Kundu, M. P. Harmer and H. M. Chan, Microstructural Evolution of a Cu and θ-Al2O3 Composite Formed By Reduction of Delafossite CuAlO2: A HAADF-STEM Study, Cryst. Growth Des., 2016, 16, 380–385, DOI:10.1021/acs.cgd.5b01362.
- C. Linderholm, A. Lyngfelt, A. Cuadrat and E. Jerndal, Chemical-looping combustion of solid fuels – operation in a 10 kW unit with two fuels, above-bed and in-bed fuel feed and two oxygen carriers, manganese ore and ilmenite, Fuel, 2012, 102, 808–822, DOI:10.1016/j.fuel.2012.05.010.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra22712k. All data accompanying this publication are directly available within the publication or within the accompanying ESI; raw datafiles are available at http://https://www.repository.cam.ac.uk; https://doi.org/10.17863/CAM.6486 |
| ‡ Present address: Merz Court, School of Chemical Engineering and Advanced Materials, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. |
| This journal is © The Royal Society of Chemistry 2016 |