
Green fabrication of magnetic recoverable graphene/MnFe2O4 hybrids for efficient decomposition of methylene blue and the Mn/Fe redox synergetic mechanism†
Xiyue Penga,
Jiangying Qu*ab,
Shuo Tiana,
Yanwei Dinga,
Xi Haia,
Bo Jiangc,
Mingbo Wu *c and
Jieshan Qiub
*c and
Jieshan Qiub
aFaculty of Chemistry and Chemical Engineering, Liaoning Normal University, Dalian, 116029, China. E-mail: qujy@lnnu.edu.cn; Tel: +86-411-82158329
bCarbon Research Laboratory, Center for Nano Materials and Science, School of Chemical Engineering, State Key Lab of Fine Chemicals, Dalian University of Technology, Dalian, 116024, China
cState Key Laboratory of Heavy Oil Processing, School of Chemical Engineering, China University of Petroleum, Qingdao, 266580, China. E-mail: wumb@upc.edu.cn; Tel: +86-532-86983452
First published on 27th October 2016
Abstract
Herein, we report an environmentally benign synthesis of a high-performance reduced graphene oxide/MnFe2O4 (RGO/MnFe2O4) catalyst for methylene blue (MB) decomposition in neutral solution using a GO/MnSO4 suspension from a modified Hummers method and FeSO4 as the precursors. The as-prepared RGO/MnFe2O4 catalyst shows exceptional performance towards the MB decomposition in the presence of H2O2. In particular, 10 mL of MB (50 mg L−1) can be thoroughly decolorized in 130 min and 78% mineralized with 5 mg of RGO/MnFe2O4 hybrid at room temperature. More interestingly, the catalysts can be magnetically recycled. The good catalytic performance of the RGO/MnFe2O4 hybrid is not only attributed to the synergetic effects of RGO, MnFe2O4, H2O2 and MB molecules, but also related to the redox couples of Fe/Mn ions during the reaction. We have firstly experimentally demonstrated that the catalytic performance of MnFe2O4 is dominated by Fe3+/Fe2+ in the initial stage (<70 min) then by Mn3+/Mn2+ in the later stage (>70 min), while Fe2+/Mn3+ redox in turn benefits the redox cycles of Fe3+/Fe2+ and Mn3+/Mn2+. Our results not only provide an alternative strategy for green synthesis of high-performance functional nanomaterials, but also promote a deep understanding of the mechanism of MnFe2O4 catalyst for MB decomposition.
Introduction
An ever increasing amount of toxic and hazardous waste water is being generated from industries and consequently results in serious environmental issues.1–3 Organic dyes are a major type of pollutant widely existing in nature water, and thus the development of powerful and practical methods for the degradation of organic dyes has attracted world-wide attention.4 As an effective advanced oxidation technology, catalysts employing the traditional homogeneous Fenton reaction with free Fe2+ have been widely used to decompose organic dyes. However, the narrow pH range (pH 2–3) and the polluted iron sludge generated at the end of process prohibit the practical application of this technology.5,6 To overcome these drawbacks, potential applications of heterogeneous Fenton-like catalysts have been investigated widely. Specifically, iron based Fenton-like catalysts such as Fe3O4, Fe2O3, FeOOH7–9 and manganese based ones such as MnO2, Mn3O4 (ref. 10 and 11) are two common kinds of heterogeneous catalysts for the degradation of organic dyes. Nevertheless, the catalytic performance of these mono-metal oxide catalysts is far from satisfaction. Furthermore, the recycling of these heterogeneous catalysts as another crucial issue needs to be addressed (except for Fe3O4 and γ-Fe2O3, δ-FeOOH).4 The introduction of Mn into the structure of Fe3O4 to produce magnetic Mn–Fe bimetal oxide catalyst provides new route for the enhancement of Fenton activity.12–14 In this field, MnFe2O4 particles have been intensively investigated mainly because of their high activity, good magnetization, nice biocompatibility and low cost.15,16 For example, Fang et al. synthesized coated MnFe2O4 nanocomposites for the decomposition of Red X-3B with the assistance of microwave.17 Yang et al. prepared the magnetic MnFe2O4 as the adsorbent for the removals of methylene blue (MB) and Congo red in single and binary dye systems.18 Despite these advances, the unique magnetic property of MnFe2O4 particles usually leads to their aggregation, thus decreasing their catalytic efficiency.19 As a result, extensive efforts have to be made in controlling the size and dispersion of MnFe2O4 particles for enhanced catalytic performance.Graphite and graphene are two common carbon materials for fabricating nanocomposites.20,21 The former exhibits significant potential in the improvement of thermal conductivities of the composites.22,23 However, the later with unique electronic property, large surface area, high chemical stability and good mechanical flexibility can not only serve as an ideal substrate for growing functional nanoparticles, but also as a charge transfer medium in the catalytic reactions.24–26 Nowadays, it is demonstrated that magnetic heterogeneous catalysts can be uniformly dispersed on graphene to fabricate the hybrids towards the degradation of water pollution.27 For example, Bai et al. fabricated graphene/MnFe2O4 hybrids from GO, Fe2+ and Mn2+, which could remove over 75% rhodamine B and 95% MB within 180 min.28 Using the similar method, Yao's group demonstrated that magnetic graphene/MnFe2O4 exhibited 97% degradation of MB in 120 min with the assistance of peroxymonosulfate, which was much higher than that of bare MnFe2O4.29 These studies indicate that the combination of graphene with magnetic MnFe2O4 provides the potential catalytic route which meets the requirements of high catalytic activity, easy recovery and reuse. However, the common methods for the fabrication of graphene/MnFe2O4 catalysts often begin with GO as the precursor and additional Mn2+ ions as Mn source. Generally, GO produced by the modified Hummers method requires intercalation and oxidation of graphite with a stoichiometric amount of KMnO4 and H2SO4 and results in a large amount of waste containing H+, Mn2+ and SO42− ions.30,31 For the viewpoint of the whole synthesis, the production of RGO/MnFe2O4 will be more valued if the H+, Mn2+ and SO42− ions can be fully utilized, which will not only avoid the tedious purification process of GO as well as the production of large amount of waste, but also contribute to highly atom-economic synthesis.32 We previously fabricated reduced graphene oxide/MnO2 (RGO/MnO2) and GO/Mn3O4 composites with GO/MnSO4 suspension as Mn source and the resultant samples exhibited good catalytic performances for 100% degradation of MB in 5 min at 50 °C and 200 min at room temperature, respectively.31,32 In spite of our efforts, the enhanced catalytic activity, facile recycling treatment and further understanding of the catalytic mechanism are desired.
It is widely reported that hydroxyl radical (˙OH) radical is crucial parameter for understanding the effective degradation of dyes with H2O2 assisted heterogeneous Fenton-like reaction.12 In principle, H2O2 activation mechanism is critically depends on the nature of the catalyst. The mono-metal catalyst such as iron oxide, manganese oxide with multiple redox states to activate H2O2 into ˙OH has been widely studied.15,29 However, the exact role of oxidation state of different components for bimetal oxide catalyst during Fenton reaction needs to be further explored based on the experimental data.
In this work, we further develop environmentally benign synthesis of magnetic RGO/MnFe2O4 hybrids from the pristine suspension (GO/MnSO4) with the addition of FeSO4 as the iron source. The work provides the effective utilization of Mn2+ ions in the pristine suspension of GO/MnSO4, and the introduction of RGO can facilitate the dispersion of the in situ formed magnetic particles, via which their aggregation is significantly alleviated. Due to these structural merits, the as-prepared hybrids show improved catalytic performance towards the MB decomposition in the presence of H2O2. In addition, these catalysts can be facilely recycled due to the magnetic property of the MnFe2O4 nanoparticles. More importantly, the roles of the oxidation state of Fe(III)/Mn(II) in MnFe2O4 with varied redox property (Fe3+/Fe2+, Mn3+/Mn2+ and Fe2+/Mn3+) on the catalytic performance and the involved mechanism are investigated in detail.
Experimental
Synthesis of RGO/MnFe2O4 hybrids
The procedure for the synthesis of RGO/MnFe2O4 hybrids was carried out as follows. First, 10 mL of the homogeneous GO/MnSO4 suspension (13 mg mL−1 GO and 0.22 mol L−1 MnSO4), which was synthesized by modified Hummers method according to ref. 32 and 33, was sonicated for 20 min. Then 1.22 g FeSO4·7H2O was added and the mixed suspension was stirred at room temperature. Subsequently, 2 mol L−1 KOH was added into the above solution for another 0.5 h under bubbling air and stirring until the pH = 12. The system was then transferred in a 20 mL Teflon-lined autoclave at 150 °C for 15 h. Finally, the black precipitate (RGO/MnFe2O4) was collected by filtration, washed thoroughly with distilled water, and fully dried at 80 °C over night.RGO/MnFe2O4 hybrids with different mass contents of MnFe2O4 were synthesized by tailoring the ratio of GO/MnSO4 to FeSO4. First, GO/MnSO4 suspensions with different mass ratios of GO to MnSO4 were prepared according to ref. 33. Then, a certain amount of FeSO4·7H2O was added into the above suspension based on the theoretic molar ratio of Fe2+ to Mn2+ in the MnFe2O4 molecule, which was accurately equal to be 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The following steps by adding KOH and other procedures are similar to the above process. The resultant samples were named as RGO/MnFe2O4-X, where X represented the mass percentage of MnFe2O4 in RGO/MnFe2O4 hybrids. For comparison, RGO or bare MnFe2O4 particle were prepared in a similar way in the absence of MnFe2O4 or GO.
:1. The following steps by adding KOH and other procedures are similar to the above process. The resultant samples were named as RGO/MnFe2O4-X, where X represented the mass percentage of MnFe2O4 in RGO/MnFe2O4 hybrids. For comparison, RGO or bare MnFe2O4 particle were prepared in a similar way in the absence of MnFe2O4 or GO.
Test of the catalytic performance
The activities of RGO/MnFe2O4 hybrids for the decomposition of MB dye were investigated. In a typical test, 5 mg of the resulting catalyst and a solution which contained 10 mL of MB dye solution (50 mg L−1) as well as 5 mL of H2O2 (30 wt%) solution were mixed under continuous stirring. The concentration of MB dye was determined by UV-vis spectroscopy at 664 nm. Besides, the catalyst was magnetically separated and reused in a fresh MB and H2O2 solution after completion of the reaction for the stability tests of RGO/MnFe2O4-75. Furthermore, the formation of active ˙OH upon irradiation was chosen to evaluate the catalytic properties of the RGO/MnFe2O4 hybrids by using terephthalic acid (TA) as the probe molecule. The detailed process of catalytic activity test and the ˙OH analysis were shown in ESI Sp1 and Sp2,† respectively.The structure analyses and catalytic measurements of the obtained samples were similar to our previous report.32
Results and discussion
The characterization of RGO/MnFe2O4 hybrids
The environmentally benign synthesis of magnetic RGO/MnFe2O4 catalyst for the decomposition of MB is illustrated in Fig. 1. Firstly, we took full use of the pristine GO/MnSO4 suspension (13 mg mL−1 GO and 0.22 mol L−1 MnSO4) derived from a modified Hummers method,34,35 in which Mn2+ ions preferably combined with the O atoms of the negatively charged surface of GO sheets via electrostatic interaction.36 Additional FeSO4 and KOH were successively added into the system. After being hydrothermally treated at 150 °C for 15 h, MnFe2O4 was formed (2Mn2+ + 12OH− + 4Fe2+ + O2 = 2MnFe2O4 + 6H2O) and anchored firmly on the RGO surface to form RGO/MnFe2O4 hybrids. Moreover, the only by-product, namely, K2SO4, could be recycled for other purposes (ESI Sp3†). The resultant RGO/MnFe2O4 hybrids showed effective catalytic decomposition of MB and could be magnetically recovered. | ||
| Fig. 1 Schematic synthesis of magnetically recoverable RGO/MnFe2O4 hybrids for the decomposition of MB dye. | ||
The powder X-ray diffraction (XRD) patterns of RGO/MnFe2O4-75, bare MnFe2O4 particles and RGO were shown in Fig. 2a. MnFe2O4 particles exhibit the intense peaks corresponding to (220), (311), (400), (511) and (440) planes, which match well with the standard MnFe2O4 (JCPDS 38-0430).17 For RGO/MnFe2O4-75, the intense peaks are weaker than those of bare MnFe2O4, which may be attributed to the small size and good distribution of MnFe2O4 in the hybrid. Furthermore, RGO/MnFe2O4-75 and RGO show the broad and weak peak at around 2θ = 25° as the result of the hydrothermal reduction of GO to RGO.29
 | ||
| Fig. 2 (a) XRD patterns and (b) Raman spectra of RGO/MnFe2O4-75, MnFe2O4, and RGO. | ||
The Raman spectra of the obtained samples were shown in Fig. 2b. It is well known that the G band at 1596 cm−1 is usually assigned to sp2 carbon domains, while the D band located at 1345 cm−1 is associated with the defects and disorder carbon in the graphitic layers.31,36 The intensity ratio of D band to G band (ID/IG) is usually used as the measure of the ordering quality of carbon materials. A lower intensity ID/IG ratio of RGO/MnFe2O4-75 hybrid, i.e., 1.11 is observed compared with 1.34 of RGO, suggesting that MnFe2O4 in RGO/MnFe2O4-75 can help to increase the size of the sp2 domains. In addition, the characteristic peak at 612 cm−1 of RGO/MnFe2O4-75 matches completely with that of bare MnFe2O4 particles.29,37
XPS analysis was further employed to elaborate the surface chemical bonding states of the RGO/MnFe2O4 hybrid. The full-scale XPS spectra with C 1s, O 1s, Mn 2p, and Fe 2p spectra were shown in Fig. 3a. The spectrum of Fe 2p (Fig. 3b) has two main peaks at binding energies of 711.3 and 725.1 eV, which are related to Fe 2p3/2 and Fe 2p1/2, respectively. The satellite peaks at the position of 719.8 and 733.3 eV are also visible, confirming the presence of Fe3+ chemical state within MnFe2O4.29,38 As shown in Fig. 3c, it is found that the XPS spectrum of Mn 2p presents two main peaks of Mn 2p3/2 (641.2 eV) and Mn 2p1/2 (652.8 eV) together with a satellite peak (644.9 eV), which are in good agreement with the Mn2+ chemical state.39,40 The C 1s peak at 284.6 eV is commonly assigned to the elemental carbon (Fig. 3d).41 When comparing to the peaks of GO (the inset in Fig. 3d), the C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C peak becomes predominant (284.6 eV) while the peaks of C–O and CO decrease drastically. This means that GO has been reduced to RGO except for a small amount oxygen-containing groups after being hydrothermally treated, which agrees with the XRD result.
C peak becomes predominant (284.6 eV) while the peaks of C–O and CO decrease drastically. This means that GO has been reduced to RGO except for a small amount oxygen-containing groups after being hydrothermally treated, which agrees with the XRD result.
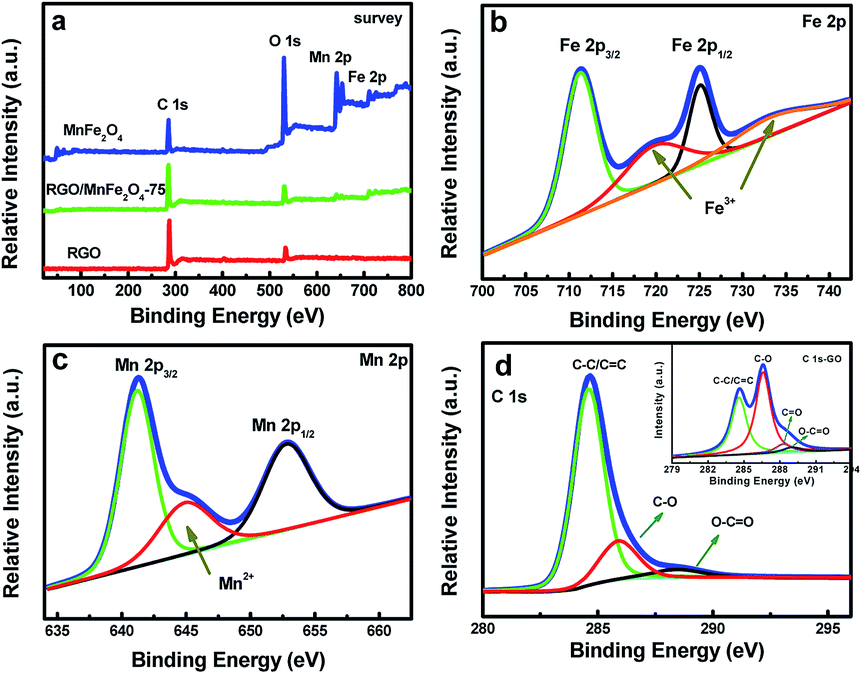 | ||
| Fig. 3 XPS spectra of RGO/MnFe2O4-75, MnFe2O4 and RGO: (a) the survey scan; (b–d) Fe 2p region, Mn 2p region and C 1s region of RGO/MnFe2O4-75; the inset in (d) is C 1s region of GO. | ||
SEM images of RGO/MnFe2O4-75 and bare MnFe2O4 were shown in Fig. 4. Sandwich-like RGO layers are decorated by MnFe2O4 spheres with the size of around 200 nm, indicating that the presence of RGO effectively inhibits the aggregation of MnFe2O4 particles (Fig. 4a and b). Meanwhile, the existence of MnFe2O4 can prevent RGO from stacking.42 Such synergistic interaction plays a positive role in the catalytic decomposition of organic pollutants due to the improved transportation of electrons between MnFe2O4 and RGO.30 For comparison, the bare MnFe2O4 particles (Fig. 4c) with diameters ranging from 100 to 500 nm seriously aggregated in the absence of RGO.
 | ||
| Fig. 4 SEM images of (a and b) RGO/MnFe2O4-75 and (c) the bare MnFe2O4. | ||
TGA was applied from room temperature to 700 °C in air flow to analyze the thermal stability and quantified composition of RGO/MnFe2O4 hybrids. As shown in Fig. 5a, the gradual weight loss in RGO/MnFe2O4-75 hybrid occurs at 20–500 °C, which is corresponding to the loss of the physically adsorbed water and the burning of RGO.31 At last, MnFe2O4 with the mass percent of 75% is left after RGO is fully burned up at 700 °C (evidenced by XRD analysis in Fig. 5b). Therefore, it is calculated that the content of MnFe2O4 in RGO/MnFe2O4 is 75%. The mass ratios of MnFe2O4 in RGO/MnFe2O4 with 80% and 55% can be further tuned by adjusting the weight ratio of pristine GO/MnSO4 suspension to FeSO4 (shown in Experimental Section).
 | ||
| Fig. 5 (a) TGA curves of RGO/MnFe2O4 hybrids with different MnFe2O4 contents and (b) XRD pattern of residual MnFe2O4 after the burning of RGO of RGO/MnFe2O4-75. | ||
Catalytic activities and stability test of RGO/MnFe2O4 hybrids
The catalytic activities of the as-prepared samples were evaluated at a given reaction interval in the presence of H2O2. The UV-vis absorption spectra of MB solution treated by RGO/MnFe2O4-75 hybrid were shown in Fig. 6a. The two characteristic absorption peaks appearing at 614 and 664 nm are contributed to the typical MB peaks in visible region at the starting solution.43 After adding both RGO/MnFe2O4-75 and H2O2, the absorption peaks of MB solution obviously decrease. It can be seen that the absorption of MB is relatively rapid in the initial 50 min and its gradual blue shift is observed with prolonged reaction time, indicating the breakdown of chromophore structure of MB.32 As a result, the solution turns colorless gradually within 130 min and the corresponding photo images were shown in the inset of Fig. 6a. A series of comparative experiments were designed to verify the catalytic performance of the obtained samples. Fig. 6b shows the result of the decomposition degree of MB dye within 130 min for different samples. For comparison, the degree of MB decolorization is 21% without catalyst (only MB and H2O2), which is slightly higher than 17.3% in the absence of H2O2 (only MB and RGO/MnFe2O4-75) because of the low specific surface area (29 m2 g−1) of RGO/MnFe2O4-75 hybrid. Therefore, the synergetic effect of catalyst and H2O2 plays an important role in efficient decomposition of MB dye from water. To further explore the synergistic effect of MnFe2O4 and RGO in RGO/MnFe2O4 hybrids, the catalytic activities of RGO/MnFe2O4 with different MnFe2O4 contents as well as pure RGO in the decomposition of MB were compared. It is found that 27% and 24% of MB are decolorized by bare MnFe2O4 particles and RGO, respectively. As the mass ratio of MnFe2O4 in RGO/MnFe2O4 hybrids varies from 55 to 75 wt%, the decolorization degree of MB increases from 71.2 to 100% within 130 min, which is more effective than that of the reported RGO/MnFe2O4 hybrid (95% MB decomposition within 180 min)28 and our previous work on RGO/MnO2 and GO/Mn3O4 compositions (100% MB decomposition within 5 min at 50 °C and 200 min at room temperature, respectively).31,32 The further increased MnFe2O4 content to 80 wt%, however, results in a low MB decolorization rate of 82% under the same condition. Thus, 75 wt% is the best content for MnFe2O4 in RGO/MnFe2O4 hybrid for MB decolorization. The mechanism of the significant activity enhancement of RGO/MnFe2O4 hybrids is illustrated as below. First, RGO can prevent the aggregation of MnFe2O4 particles as well as serve as the electron transfer bridges to enhance the MB decomposition based on the XRD, Raman and SEM analyses. Considering 2D planar structure and giant π-conjugation system of RGO, the MB molecules can be adsorbed on the surface of RGO and retained in close proximity to the active sites of MnFe2O4, thus providing great opportunities to degrade the contaminants.44 Second, the uniformly dispersed MnFe2O4 particles can increase the number of active sites for the catalytic decomposition of MB as well. Besides, Mn2+ also plays an important role in accelerating the electron transfer process between Fe2+/Fe3+, and thus improves the catalytic activity to some extent,45,46 which will be discussed in the following part.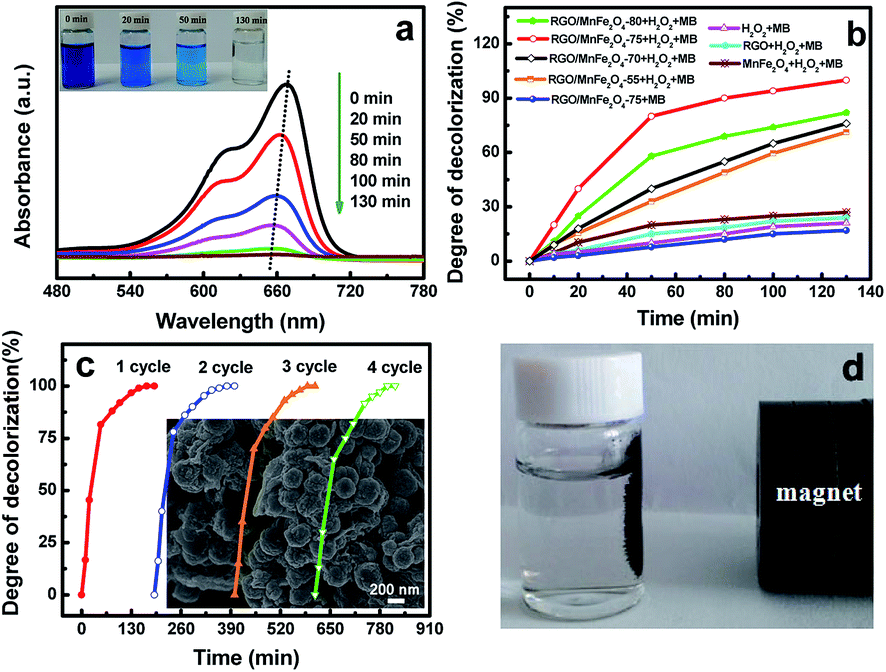 | ||
| Fig. 6 (a) Absorption spectra of MB solution (50 mg L−1, 10 mL) of RGO/MnFe2O4-75 hybrid in different time intervals (the inset is the photo image); (b) time profiles of MB decomposition under different conditions; (c) the decomposition of MB with RGO/MnFe2O4-R, the inset is its SEM image; (d) the photo image of magnetically recyclable RGO/MnFe2O4. | ||
Undoubtedly, the stability of the catalyst is quite important for practical applications. Thus, the stability test of RGO/MnFe2O4-75 is shown in Fig. 6c. Four recycling runs of RGO/MnFe2O4-75 were performed without any noticeable loss of catalytic ability. The catalyst named as RGO/MnFe2O4-75-R which was recovered after the test was further studied by SEM (the inset of Fig. 6c) and XRD analyses (Fig. 2a), which indicates that the size, morphology, and structure of the catalysts remains almost unchanged, further demonstrating their excellent stability. The concentration of the slightly leached Mn ions (about 0.5 wt%) from RGO/MnFe2O4-75 was further detected by inductive coupled plasma emission spectrometer (ICP),32 indicating the excellent stability of RGO/MnFe2O4-75 hybrid. It's worth mentioning that the catalyst can be completely recovered with a permanent magnet after each run (as shown in Fig. 6d), which not only simplifies operation steps and improves working efficiency, but also meets the demands of green chemistry.
As is known to all, the SO42− mineralization is widely used to evaluate the MB degradation efficiency.31 In this work, the SO42− concentration in the final solution was measured to be 7.1 mg L−1 (0.4 mg L−1 of the SO42− comes from the H2O2 reagent). According to the theoretically calculated concentration (about 8.5 mg L−1) after 100% decolorization of MB by RGO/MnFe2O4-75, we draw the conclusion that MB dye has been effectively decolorized and about 79% has been mineralized over RGO/MnFe2O4-75 hybrid, which is higher than those of previously reported GO/Mn3O4 composite (77%),32 RGO/MnO2 composite (66%)31 and Mn3O4/graphene hybrid (27%).36
Possible catalytic mechanism
In order to deeply understand the catalytic performance of as-prepared samples, the cumulative amount of ˙OH radicals was measured by photoluminescence (PL) technique with the similar method according to our previous work.32 The TA solution was selected as a probe molecule to react with ˙OH to produce highly fluorescent product 2-hydroxyterephthalic acid.10 It can be observed from Fig. 7a that a gradual increase of the PL intensity appears at 445 nm under UV light irradiation with the reaction time, suggesting that fluorescence presents because of the reactions of TA with ˙OH, showing a proportional relationship between PL intensity and the amount of generated ˙OH.32 Fig. 7b presents a comparison of the PL intensities of RGO/MnFe2O4-75, MnFe2O4 and RGO samples in the assistance of H2O2. As shown in Fig. 6b, the amount of produced ˙OH radicals by the tested samples decreases in the same order of their MB catalytic performances (RGO/MnFe2O4-75 > MnFe2O4 > RGO). This agreement between catalytic performance and hydroxyl radical measurement further clarifies that positive effect between MnFe2O4 and RGO on the MB decomposition in this study. | ||
| Fig. 7 (a) PL spectra of 5 × 10−4 M basic TA solution treated by RGO/MnFe2O4-75 irradiated by UV light under different time (excitation at 325 nm) and (b) PL spectra of 5 × 10−4 M basic TA solution treated by various samples irradiated by UV light at 50 min. | ||
The mechanism of the enhanced catalytic performance for RGO/MnFe2O4 may be explained by the possible electron transfer process of MnFe2O4 during the reaction. According to the experimental results of XPS analysis, iron and manganese of MnFe2O4 exist mainly in the oxidation state, i.e. Fe(III) and Mn(II), respectively. Thus, Fe2O3 and MnO samples were prepared for further analysis (as shown in Sp4†). The catalytic performances of individual Fe2O3, MnO and MnFe2O4 were evaluated under the same condition (Fig. 8a). It is observed that MB decomposition rate is dominated by Fe2O3 in the initial stage (<70 min) then by MnO in the later stage (>70 min). As a result, the performance of MnFe2O4 is superior to that of individual MnO and Fe2O3 during the whole process. The competitive PL intensities of MnO and Fe2O3 are shown in Fig. 8b–d. The results demonstrate that the performance of above catalysts is related to the amount of ˙OH. In the starting period (20 min), the amount of ˙OH produced by Fe2O3 is more than that of MnO (as shown in Fig. 8b), which exhibits a contrary result when the reaction time is more than 70 min (Fig. 8c and d). The amount of ˙OH produced by MnFe2O4 is larger than those of MnO and Fe2O3 in the whole process, which are in good agreement with their catalytic performances as described in Fig. 8a.
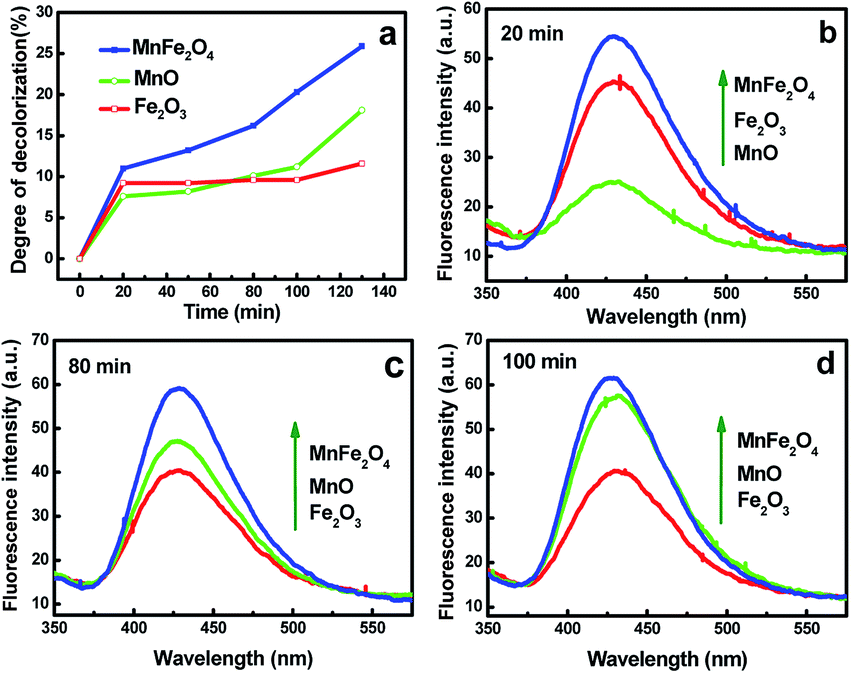 | ||
| Fig. 8 (a) Time profiles of MB decolorization (50 mg L−1, 10 mL) treated by different samples under the same condition; (b–d) PL spectra of various samples in a 5 × 10−4 M basic TA solution under UV light irradiation. | ||
The performance of Fe2O3 may be attributed to the significant effect of Fe3+/Fe2+ cycling to yield ˙OH radicals for the degradation of organic pollutants. At first, Fe3+ is reduced to Fe2+ which subsequently reacts with H2O2 to produce ˙OH and Fe3+. Consequently, such process accelerates the Fe3+/Fe2+ cycle to form the large amount of strong oxidant ˙OH for the degradation of MB.27,47 The main process may be described as eqn (1) and (2).
| Fe3+ + H2O2 → Fe2+ + ˙OOH + H+ | (1) |
| Fe2+ + H2O2 → Fe3+ + ˙OH + OH− | (2) |
The reactions between Mn2+ and H2O2 can also contribute to the generation of ˙OH, which would be responsible for the remarkable increase on the catalytic activities,46 as shown in eqn (3) and (4).
| Mn2+ + H2O2 → Mn3+ + ˙OH + OH− | (3) |
| Mn3+ + H2O2 → Mn2+ + ˙OOH + H+ | (4) |
Beside the Fe3+/Fe2+ and Mn3+/Mn2+ cycles, the reaction between Fe2+ and Mn3+ is thermodynamically favorable, which benefits the redox cycles of Fe3+/Fe2+ (Eθ(Fe3+/Fe2+) = 0.77 V) and Mn3+/Mn2+ (Eθ(Mn3+/Mn2+) = 1.51 V) (as shown in eqn (5)).
| Fe2+ + Mn3+ → Fe3+ + Mn2+, Eθ = 0.74 V | (5) |
Given the better catalytic performance of MnFe2O4 than those of individual Fe2O3 and MnO, it is anticipated that the synergetic effects of the redox couples of Mn3+/Mn2+, Fe3+/Fe2+ and Fe2+/Mn3+ during the reaction is responsible for the enhanced performance.
In order to further demonstrate the oxidation state of Mn and Fe of MnFe2O4 during the reaction, XPS spectra of the cycled RGO/MnFe2O4-75-R were further recorded (see Sp5†). Comparing to the Mn 2p and Fe 2p in Fig. 3b and c, the RGO/MnFe2O4-75-R hybrid exists the same peaks with the fresh one. However, there are some slight differences of the binding energies and the full widths at half maximum (FWHM) values of Fe 2p as well as Mn 2p before and after Fenton reaction, which have been displayed in Tables S1 and S2,† respectively. Such results may be attributed to the electron transfer of Fe3+/Fe2+ and Mn3+/Mn2+ in the catalytic process. H2-TPR measurement was further applied in the MnFe2O4 catalyst with Mn3O4 and Fe2O3 as the references (Sp6†). It is found that the reduction step of Fe3+ to Fe2+ starts at a higher temperature (320 °C) comparing with that of Mn3+ to Mn2+ (210 °C).48 This observation suggests that Mn3+/Mn2+ presents higher oxidation capacity than Fe3+/Fe2+, which is in agreement the reaction described in eqn (5).
Accordingly, the mechanism schematic of MB decomposition on RGO/MnFe2O4 catalyst with the assistance of H2O2 was shown in Fig. 9. At first, MB molecules are adsorbed on the RGO and its giant π-conjugation system and 2D planar structure help to promote the electron transfer during the catalytic reaction.32,49 Besides, MnFe2O4 nanoparticles anchored on the RGO surface catalyze H2O2 to produce large amount of ˙OH radicals and constantly facilitate the decomposition of MB molecules into CO2, H2O and SO42− etc. During the catalytic process, the coexistence of the internal electron transfer in Fe3+/Fe2+ and Mn3+/Mn2+ redox couples have prominent contributions to the significant catalytic performance, wherein the former help to that of the earlier 70 min and the later plays a positive role in the last period. Furthermore, the reduction of Mn3+ by Fe2+ during the reaction is thermodynamically favorable, which in turn benefits the redox cycles of Fe3+/Fe2+ and Mn3+/Mn2+. Therefore, the coupling contributions of RGO/MnFe2O4 catalyst result in an appreciable improvement in the decomposition of MB.
 | ||
| Fig. 9 Schematic of the mechanism of MB decomposition on RGO/MnFe2O4 hybrid with the assistance of H2O2. | ||
Conclusions
In summary, we have successfully synthesized magnetic RGO/MnFe2O4 hybrids with pristine GO/MnSO4 suspension derived from a modified Hummers method and ferrous sulfate as the main precursors. Such a process not only alleviates the tedious purification process for GO production, but also contributes to a high atom-economic synthesis. The combination of magnetic MnFe2O4 particles with RGO plays an important role in controlling the size and distribution of the MnFe2O4 nanoparticles, thus allowing excellent catalytic performance for the decomposition of MB. In particular, MB dye molecules in neutral solution can be fully decomposed within 130 min in the presence of H2O2 at room temperature. The sequential synergetic actions of Fe3+/Fe2+, Mn3+/Mn2+ and Fe2+/Mn3+ redox couples at different stages contribute to the excellent catalytic activity. Besides, the magnetic property of the MnFe2O4 nanoparticles greatly simplifies the recycling performance. This work not only offers a new approach to fabricate graphene-based functional hybrids, but also benefits understanding the mechanism of transition metals on the heterogeneous Fenton system.Acknowledgements
This work is supported by the NSFC (No. 51372277, 50902066), China Postdoctoral Science Foundation (2013M530922, 2014T70253), Program for Liaoning Excellent Talents in University (LJQ2014118), the Fundamental Research Funds for the Central Universities (No. 15CX08005A), Natural Science Fund of Liaoning Province (201602458).References
- N. N. Tušar, D. Maučec, M. Rangus, I. Arčon, M. Mazaj, M. Cotman, A. Pintar and V. Kaučič, Adv. Funct. Mater., 2012, 22, 820–826 CrossRef.
- B. Jiang, Y. Liu, J. Zheng, M. Tan, Z. Wang and M. Wu, Environ. Sci. Technol., 2015, 49, 12363–12371 CrossRef CAS PubMed.
- B. Jiang, X. Wang, Y. Liu, Z. Wang, J. Zheng and M. Wu, J. Hazard. Mater., 2016, 304, 457–466 CrossRef CAS PubMed.
- M. Munoz, Z. M. de Pedro, J. A. Casas and J. J. Rodriguez, Appl. Catal., B, 2015, 176–177, 249–265 CrossRef CAS.
- S. Navalon, M. Alvaro and H. Garcia, Appl. Catal., B, 2010, 99, 1–26 CrossRef CAS.
- M. Cheng, G. Zeng, D. Huang, C. Lai, P. Xu, C. Zhang and Y. Liu, Chem. Eng. J., 2016, 284, 582–598 CrossRef CAS.
- Y. L. Pang, S. Lim, H. C. Ong and W. T. Chong, Ceram. Int., 2016, 42, 9–34 CrossRef CAS.
- T. Zeng, W. Chen, C. M. Cirtiu, A. Moores, G. Song and C. Li, Green Chem., 2010, 12, 570–573 RSC.
- B. Wang, H. Wu, L. Yu, R. Xu, T. T. Lim and X. W. Lou, Adv. Mater., 2012, 24, 1111–1116 CrossRef CAS PubMed.
- L. Zhang, Y. Nie, C. Hu and X. Hu, J. Hazard. Mater., 2011, 190, 780–785 CrossRef CAS PubMed.
- T. Rhadfi, J. Y. Piquemal, L. Sicard, F. Herbst, E. Briot, M. Benedetti and A. Atlamsani, Appl. Catal., A, 2010, 386, 132–139 CrossRef CAS.
- Y. Zhong, X. Liang, Z. He, W. Tan, J. Zhu, P. Yuan, R. Zhu and H. He, Appl. Catal., B, 2014, 150–151, 612–618 CrossRef CAS.
- Y. Guo, L. Zhang, X. Liu, B. Li, D. Tang, W. Liu and W. Qin, J. Mater. Chem. A, 2016, 4, 4044–4055 CAS.
- H. Z. P. M. Rice, S. X. Wang and S. Sun, J. Am. Chem. Soc., 2004, 126, 11458–11459 CrossRef PubMed.
- P. Deng, F. Mou, X. Li, Z. Deng, J. Sun, L. Xu and J. Guan, J. Mater. Chem. A, 2016, 4, 11768–11774 Search PubMed.
- D. Vilela, J. Parmar, Y. Zeng, Y. Zhao and S. Sanchez, Nano Lett., 2016, 16, 2860–2866 CrossRef CAS PubMed.
- X. Fang, J. Xiao, S. Yang, H. He and C. Sun, Appl. Catal., B, 2015, 162, 544–550 CrossRef CAS.
- L. Yang, Y. Zhang, X. Liu, X. Jiang, Z. Zhang, T. Zhang and L. Zhang, Chem. Eng. J., 2014, 246, 88–96 CrossRef CAS.
- G. Chen, J. Wang, L. Zhou, W. Ma, D. Zhang, F. Ren, H. Yan, G. Qiu, X. Liu and P. Joy, J. Am. Ceram. Soc., 2012, 95, 3569–3576 CrossRef CAS.
- J. Gu, X. Yang, Z. Lv, N. Li, C. Liang and Q. Zhang, Int. J. Heat Mass Transfer, 2016, 92, 15–22 CrossRef CAS.
- H. Gu, C. Ma, J. Gu, J. Guo, X. Yan, J. Huang, Q. Zhang and Z. Guo, J. Mater. Chem. C, 2016, 4, 5890–5906 RSC.
- J. Gu, N. Li, L. Tian, Z. Lv and Q. Zhang, RSC Adv., 2015, 5, 36334–36339 RSC.
- J. Gu, C. Xie, H. Li, J. Dang, W. Geng and Q. Zhang, Polym. Compos., 2013, 35, 1087–1092 Search PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- B. Xia, Y. Yan, X. Wang and X. W. Lou, Mater. Horiz., 2014, 1, 379–399 RSC.
- H. Hu, Z. Zhao, Y. Gogotsi and J. Qiu, Environ. Sci. Technol. Lett., 2014, 1, 214–220 CrossRef CAS.
- S. Han, L. Hu, Z. Liang, S. Wageh, A. A. Al-Ghamdi, Y. Chen and X. Fang, Adv. Funct. Mater., 2014, 24, 5719–5727 CrossRef CAS.
- S. Bai, X. Shen, X. Zhong, Y. Liu, G. Zhu, X. Xu and K. Chen, Carbon, 2012, 50, 2337–2346 CrossRef CAS.
- Y. Yao, Y. Cai, F. Lu, F. Wei, X. Wang and S. Wang, J. Hazard. Mater., 2014, 270, 61–70 CrossRef CAS PubMed.
- W. Zhao, J. Kong, H. Liu, Q. Zhuang, J. Gu and Z. Guo, Nanoscale, 2016 10.1039/c6nr06622d.
- J. Qu, L. Shi, C. He, F. Gao, B. Li, Q. Zhou, H. Hu, G. Shao, X. Wang and J. Qiu, Carbon, 2014, 66, 485–492 CrossRef CAS.
- Y. Li, J. Qu, F. Gao, S. Lv, L. Shi, C. He and J. Sun, Appl. Catal., B, 2015, 162, 268–274 CrossRef CAS.
- J. Qu, F. Gao, Q. Zhou, Z. Wang, H. Hu, B. Li, W. Wan, X. Wang and J. Qiu, Nanoscale, 2013, 5, 2999–3005 RSC.
- L. L. Peng, X. Peng, B. R. Liu, C. Z. Wu, Y. Xie and G. H. Yu, Nano Lett., 2013, 13, 2151–2157 CrossRef CAS PubMed.
- H. Hu, Z. Zhao, Q. Zhou, Y. Gogotsi and J. Qiu, Carbon, 2012, 50, 3267–3273 CrossRef CAS.
- N. Li, Z. F. Geng, M. H. Cao, L. Ren, X. Y. Zhao, B. Liu, Y. Tian and C. W. Hu, Carbon, 2013, 54, 124–132 CrossRef CAS.
- Y. L. Xiao, J. T. Zai, L. Q. Tao, B. Li, Q. Y. Han, C. Yu and X. F. Qian, Phys. Chem. Chem. Phys., 2013, 15, 3939–3945 RSC.
- D. C. Hong, Y. Yamada, T. Nagatomi, Y. Takai and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 19572–19575 CrossRef CAS PubMed.
- Y. S. Fu, P. Xiong, H. Q. Chen, X. Q. Sun and X. Wang, Ind. Eng. Chem. Res., 2012, 51, 725–731 CrossRef.
- Y. Zhou, B. Xiao, S. Q. Liu, Z. Meng, Z. G. Chen, C. Y. Zou, C. B. Liu, F. Chen and X. Zhou, Chem. Eng. J., 2016, 283, 266–275 CrossRef CAS.
- J. Gu, C. Liang, J. Dang, W. Dong and Q. Zhang, RSC Adv., 2016, 6, 35809–35814 RSC.
- J. Zhu, S. Wei, H. Gu, S. B. Rapole, Q. Wang, Z. Luo, N. Haldolaarachchige, D. P. Young and Z. Guo, Environ. Sci. Technol., 2012, 46(2), 977–985 CrossRef CAS PubMed.
- Z. Bai, B. Sun, N. Fan, Z. Ju, M. Li, L. Xu and Y. Qian, Chem.–Eur. J., 2012, 18, 5319–5324 CrossRef CAS PubMed.
- J. Chun, H. Lee, S. H. Lee, S. W. Hong, J. Lee, C. Lee and J. Lee, Chemosphere, 2012, 89, 1230–1237 CrossRef CAS PubMed.
- R. C. Costa, M. F. Lelis, L. C. Oliveira, J. D. Fabris, J. D. Ardisson, R. R. Rios, C. N. Silva and R. M. Lago, J. Hazard. Mater., 2006, 129, 171–178 CrossRef CAS PubMed.
- R. C. Costa, M. F. Lelis, L. C. Oliveira, J. D. Fabris, J. D. Ardisson, R. R. Rios, C. N. Silva and R. M. Lago, Catal. Commun., 2003, 4, 525–529 CrossRef CAS.
- S. Guo, G. Zhang, Y. Guo and J. C. Yu, Carbon, 2013, 60, 437–444 CrossRef CAS.
- Y. Wang, H. Zhao, M. Li, J. Fan and G. Zhao, Appl. Catal., B, 2014, 147, 534–545 CrossRef CAS.
- L. Zhang, Q. Zhang, H. Xie, J. Guo, H. Lyu, Y. Li, Z. Sun, H. Wang and Z. Guo, Appl. Catal., B, 2017, 201, 470–478 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24320g |
| This journal is © The Royal Society of Chemistry 2016 |