
Cyclodextrin grafted calcium carbonate vaterite particles: efficient system for tailored release of hydrophobic anticancer or hormone drugs
Jaya R. Lakkakula*abc,
Rajendra Kurapati d,
Ivan Tyngae,
Heidi Abrahamsee,
Ashok M. Raichur*ad and
Rui Werner Maçedo Krause*b
d,
Ivan Tyngae,
Heidi Abrahamsee,
Ashok M. Raichur*ad and
Rui Werner Maçedo Krause*b
aDepartment of Applied Chemistry, Center for Nanomaterials Science, University of Johannesburg, Doornfontein 2028, South Africa
bDepartment of Chemistry, Rhodes University, Grahamstown 6140, South Africa
cDepartment of Bioscience and Bioengineering, IIT Bombay, Powai, Mumbai, India. E-mail: spencerjaya@gmail.com; Tel: +91 7506 356 388
dDepartment of Materials Engineering, Indian Institute of Science, Bangalore, 560 012, India
eLaser Research Centre, Faculty of Health Sciences, University of Johannesburg, Doornfontein 2028, South Africa
First published on 26th October 2016
Abstract
Porous CaCO3 microparticles have been used earlier for sustained drug release of hydrophilic drugs but have certain drawbacks for use with hydrophobic drugs. Hence, to overcome these drawbacks, a novel composite of CaCO3 along with cyclodextrin (CD–CaCO3) for the delivery of hydrophobic drugs was developed. Cyclodextrins (CDs), when incorporated within CaCO3, increased the porosity and surface area of microparticles thereby enhancing the encapsulation efficiency of hydrophobic drugs (5-Fluorouracil or Na-L-thyroxine) by forming inclusion complexes with cyclodextrin. Thermogravimetric and FTIR studies confirmed the interaction between the cyclodextrin and CaCO3 microparticles. Raman spectra confirmed the peak of vaterite crystals before and after loading of hydrophobic drugs within the composite. In vitro release studies when performed at pH 4.8 (5-Fu) and pH 1.2 (Na-L-thy) showed release at low pH as CaCO3 is soluble at acidic pH unlike slower release at basic pH. Release kinetics followed a Higuchi kinetic model at pH 4.8 (5-Fu) and pH 1.2 (Na-L-thy) respectively.
1. Introduction
One of the major challenges being faced by the pharmaceutical industry is the development of efficient drug carriers for the delivery of hydrophobic drugs. Drug delivery systems (DDS) play an important role by enhancing the efficacy of drugs, their biodistribution and bioavailability. Various delivery systems have been developed to encapsulate hydrophobic drugs, such as hydrogel polymer micelles, membrane lipids, dendrimers,1 liposomes, polymeric micro/nanoparticles etc.2–4 Most of these systems have limited usage because of instability, toxicity, undesirable release profiles, the requirement of high salt concentrations, low loading capacity and poor biodegradability.5Recently, porous inorganic mesoporous particles such as silica, calcium carbonate and calcium phosphate have emerged as potential drug delivery systems for hydrophobic drugs. These DDS have also been extensively studied for biomedical applications like gene delivery, biomedical analysis and bioimaging. Inorganic porous materials are favoured over other existing DDS in many areas, since they offer higher drug encapsulation irrespective of the nature of the drugs, high mechanical and chemical stability under physiological conditions,5 good biocompatibility and ease of synthesis even under scale up.6 CaCO3 is considered as one of the best drug carriers not only for the reasons listed above, but also for its biodegradability. Microparticles can be prepared by simple mixing of the Ca2+ and CO32− precursors in a co-precipitation method without any toxic organic solvents unlike silica particles. Still, research on the application and modification of CaCO3 has not been fully explored.7 In particular there is a lot of room to explore CaCO3 microparticles in combination with additives or other materials such as polymers, surfactants, liposomes and micelles.8,9 The CaCO3 porous particles are soluble at mildly acidic pH and hence the delivery system can be triggered to release the encapsulated drug at low physiological pH, which is generally favourable for drug release in cancer cells.10,11
CaCO3 mainly exists in different polymorphs namely, calcite, aragonite and vaterite. Calcite is the most stable phase whereas aragonite and vaterite are mostly metastable and readily transforms into stable state over time. Vaterite is the least stable state; it recrystallizes in water to form calcite.12,13 An ability to form stable porous microparticles with large surface areas that disintegrate easily when exposed to mild acidic conditions makes vaterite an ideal selection for drug delivery systems.11
Many efforts have been made to prepare stable vaterite or porous spherical CaCO3 particles containing additives such as proteins,14,15 dendrimers,16 surfactants,17 and charged polymer.18 In particular better porosity, morphology and particle size distribution is required. Svenskaya et al. recently demonstrated the loading efficiency of photosensitizer in vaterite micro-and nano carriers for the development of novel photodynamic therapy (PDT).19 Vaterite microspheres along with an additive, BSA have also been studied as a promising drug delivery for anticancer drug, camptothecin.20 As mentioned previously, most of the available drugs in the pharmaceutical industry suffer from hydrophobicity, lower biodistribution and agglomeration in the body fluids. Hence, there is an urgent demand for development of novel drug carriers to increase the solubility and stability of drugs for target delivery. Calcium carbonate microspheres having phospholipids molecules entrapped in the porous structure have already been reported to load hydrophobic drugs.8
Cyclodextrins (CDs) are oligosaccharides with a hydrophobic cavity inside and hydrophilic exterior. Cyclodextrins have been used as a solubilising agent to improve the solubility and stability of hydrophobic drugs. CDs can interact with molecules to form total or partial inclusion complexes with macromolecular drugs.21 The preformed inclusion complexes have been used in the formation of nanocapsules or nanoparticles using nanoprecipitation techniques.22 Inclusion complexes have also been incorporated within various polymers as delivery systems.23,24 In addition, cyclodextrins have been incorporated with polymers,25,26 micelle,27 liposomes,28 hydrogels,29 and dendrimers.30
We recently, developed a composite cyclodextrin and CaCO3 (CD–CaCO3) porous spherical micro particle system using co-precipitation and demonstrated that these hybrid porous particles are potential candidates for delivery of hydrophobic small size hydrophobic drugs using model fluorescent hydrophobic dyes such as nile red (NR) and coumarin.31 We showed the controlled release of the encapsulated hydrophobic model drugs in acidic pHs, where the drug release was triggered by the phase transformation of vaterite or porous crystals to rhombohedral or calcite crystals. However, we did not previously study the application of these hybrid porous particles for controlled delivery of therapeutic hydrophobic drugs and there in vitro cytotoxicity in mammalian cells.
5-Fu is an important chemotherapy drug, which is a pyrimidine analogue that inhibits DNA synthesis by competitive inhibition of thymidylate synthetase and therefore has a broad spectrum activity against solid tumours.32 However, delivery of 5-Fu is limited, since it has a short biological half-life (20 to 30 min) due to rapid metabolism. Complicating delivery is the fact that it has a poor oral absorption, toxic side effects on bone marrow and the gastrointestinal tract, and rapid clearance.33 Thus, extending the blood circulation time of 5-Fu will increase its efficiency, and various drug delivery system has been developed to this effect.34
On other hand, thyroxine, is secreted by thyroid gland and are essential for development of several organs including central nervous system, heart, intestine, skeletal muscle and sensory organs.35 There are several diseases related to deficiency or resistance to thyroid hormone that is an inherited endocrine human disease. Resistance to thyroid hormone is usually associated by a mutation in the TRβ gene.36 Na-L-thyroxine, is a hydrophobic drug and is associated with solubility and stability. In our previous work, formation of inclusion complex with cyclodextrin in the presence of thyroxine was demonstrated and reported.37 Microparticles have already been shown to improve the bioavailability, increase residence time and control release of drugs in the body under certain conditions. Since most DDs are designed for the delivery of a specific drug, the utilization of one DDs for different types of drugs has rarely been reported.
Thus, we extended our exploration of hybrid cyclodextrin-modified CaCO3 porous particles (CD–CaCO3) for controlled delivery of potential chemotherapeutic drugs like 5-fluorouracil (5-Fu) as well as hormone drugs like Na-L-thyroxine. In vitro drug release studies and cytotoxic assay have been studied using mammalian liver cancer cells. The hydrophobic drugs can be loaded by forming a supramolecular inclusion complex with cyclodextrin entrapped in CaCO3 particles. It provides a novel way to create a new hybrid drug delivery system.
2. Materials and methods
2.1 Materials
β-Cyclodextrin and γ-cyclodextrin was procured from Walker Chemie (Munich, Germany). Na2CO3 and CaCl2 (anhydrous), 5-fluorouracil and Na-L-thyroxine were purchased from Sigma-Aldrich (Germany) and all the chemicals were used without further purification. Deionized (DI) water was used throughout the experiment.2.2 Synthesis of CaCO3 spherical microparticles
The CaCO3 spherical microparticles were prepared using well-known co-precipitation method.38 In a typical synthesis, 0.33 M Na2CO3 was rapidly added into equal volume of 0.33 M CaCl2 solution under magnetic agitation for 2 min, the white precipitate was filtered off, similar to CD–CaCO3 particles, thoroughly washed with DI water and dried in air.2.3 Fabrication of calcium carbonate microparticles containing cyclodextrin (β or γ) CD–CaCO3
The hybrid CD–CaCO3 porous microparticles were prepared using the co-precipitation method in the presence of β or γ-cyclodextrin according to our previous method.31 Firstly, 25 mL aqueous suspension of β-CD or γ-CD (10 mg mL−1) was prepared under magnetic stirring followed by the addition of 0.33 M CaCl2 solution to the suspension of cyclodextrin to form supersaturated Ca2+–CD complex and the agitation was continued for 30 min to disperse the Ca2+–CD complex.Secondly, 25 mL of a 0.33 M Na2CO3 solution was added to the above complex resulting in the formation of a β-CD–CaCO3 or γ-CD–CaCO3 white precipitate under continuous agitation for 2 min. As prepared, hybrid particles were filtered using cellulose membrane filter (0.2 μm pore size), thoroughly washed with DI water to remove free CDs and dried for 1 h at 50 °C. β-CD–CaCO3 or γ-CD–CaCO3 was used for loading of 5-fluorouracil or Na-L-thyroxine respectively leading to the formation of β-CD–CaCO3 + 5-Fu or γ-CD–CaCO3 + Na-L-thy microparticles.
To confirm the entrapment of CDs in porous CD–CaCO3 particles, a small amount of fluorescent coumarin (5 mL of 5 mg mL−1 in ethanol) was added into the ‘Ca2+–CD’ co-chelate suspension under vigorous agitation for 25 min, followed by addition of 0.33 M CO32− solution. The co-precipitation reaction was ceased after 2 min and particles were rinsed first with ethanol–water mixture and then dried in oven at 50 °C for 1 h.
2.4 Loading of 5-fluorouracil and Na-L-thyroxine into CD–CaCO3 microparticles
The loading of 5-Fu or Na-L-thy was carried out by adding a solution of 5-Fu in DMSO (5 mg mL−1) or Na-L-thy in methanol (5 mg mL−1) to β-CD–CaCO3 or γ-CD–CaCO3, respectively and then allowing the solution to stir for 24 h. The suspensions were then centrifuged and the amounts of the loaded 5-Fu and Na-L-thy in CD–CaCO3 were determined using UV-Vis analysis. Subsequently, 5-Fu and Na-L-thy loaded microparticles were collected by centrifugation, washed with DMSO or methanol to remove the adsorbed drug on the external surface, followed by washing with chloroform and drying in an oven at 50 °C. Loading efficiency of 5-Fu and Na-L-thy was found to be approximately 20 and 10% respectively.To evaluate the amount of drug got entrapped inside CD–CaCO3 microparticles, 1 mg of 5-Fu or Na-L-thy loaded CD–CaCO3 microparticles were added separately to DMSO or methanol for 24 h. After centrifugation, the 5-Fu and Na-L-thy concentration in the supernatant was determined by UV-Vis spectroscopy (UV-Vis Shimadzu 2450) with the help of calibration curve describing the absorbance–concentration relationship.
3. Characterization techniques
3.1 Scanning electron microscopy
Scanning Electron Microscopy (SEM) was used to evaluate the morphology of CaCO3, CD–CaCO3, β-CD–CaCO3 + 5-Fu and γ-CD–CaCO3 + Na-L-thy microparticles on VEGA TESCAN at an accelerating voltage of 20 kV. Samples were dropped on a silicon wafer and dried overnight under ambient conditions. A thin layer of gold coating was applied prior to analysis.3.2 Raman spectroscopy
The Raman spectra of CaCO3, CD–CaCO3, β-CD–CaCO3 + 5-Fu and γ-CD–CaCO3 + Na-L-thy microparticles were obtained using Raman microscope (Perkin Elmer Raman Micro 200) equipped with a cool charged couple device (CCD) detector set at −50 °C and an Olympus BX51M confocal microscope. A 785 nm high-performance line-narrowed laser was used as the excitation light source. Spectra were collected over the range of 800–1200 cm−1. Background was subtracted for all the spectra.3.3 Thermogravimetric analysis
The thermal properties and stability of CaCO3 and CD–CaCO3 microparticles were assessed using a Perkin Elmer TGA (400) analyser with a heating rate of 10 °C min−1 using a temperature range of 25 to 800 °C under a nitrogen atmosphere.3.4 Brunauer–Emmett–Teller (BET) analysis
The surface area, pore size and pore volume of CaCO3 and CD–CaCO3 were calculated based on BET theory. For BET analysis, Micrometrics ASAP 2020 Surface area and Porosity Analyser software was utilised and the samples were degassed prior to the determination of the surface area.3.5 Mathematical modelling
To analyse the drug release from β-CD–CaCO3 + 5-Fu and γ-CD–CaCO3 + Na-L-thy loaded microparticles, different kinetic models (i.e. zero order, first-order, Higuchi, Hixson–Crowell cube root and Korsemeyer–Peppas model) were used to describe the release of drug from the formulations. The following graphs were plotted: cumulative % drug release versus time (zero order model), log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cumulative of % drug remaining versus time (first order kinetic model), cumulative % drug release versus square root of time (Higuchi model), logcumulative % drug release versus logtime (Korsemeyer–Peppas model) and cube root of drug% remaining versus time (Hixson–Crowell cube root law).
cumulative of % drug remaining versus time (first order kinetic model), cumulative % drug release versus square root of time (Higuchi model), logcumulative % drug release versus logtime (Korsemeyer–Peppas model) and cube root of drug% remaining versus time (Hixson–Crowell cube root law).
3.6 Cell study
The A549 cancer cell line (ATCC: HTB 53) was used throughout this study and grown to 85% confluence in 25 mL complete Roswell Park Memorial Institute (RPMI, Life Technologies, RSA, Gibco 3060301), which was supplemented with 10% (v/v) FBS (Gibco Invitrogen Corporation, 10106), 1% (v/v) penicillin–streptomycin (PAA Laboratories GmbH, P11-010) and 1 μg mL−1 amphotericin B (PAA Laboratories GmbH, P11-001) in an 85% humidified atmosphere at 37 °C and 5% CO2. For experiment purposes, A549 cells were seeded at a final concentration of 1 × 105 in 3.4 cm2 diameter culture dishes and incubated for 4 h to allow the cells to attach. The induced cellular effects of β-CD–CaCO3, 5-Fu and their combination at three different concentrations (25, 50 and 100 μM mL−1) were investigated 24 h after cell treatment.Ten microlitres of cell suspension was added to 90 μL of 0.4% trypan blue (Sigma Aldrich, T8154) in HBSS (1:10 dilution) and carefully mixed. Then ten microlitres of the mixture was transferred to both counting chambers on either side of a Bright-Line™ hemocytometer by placing a pipette tip at the edge of the coverslip and allowing each chamber to fill by capillary action. Cells were viewed using a microscope (Olympus, CKX41) and total number of cells was counted and the percentage viability was determined by multiplying the average number of cells per square by 105 (dilution factor × 104) and the fraction of the number of viable cells over the total number of cells by 100.
Fifty microliters of ATP CellTiter-Glo® reagent was added to an equal volume of cell suspension for each different treatment in an opaque-walled 96 well plate (BD Biosciences, 353296). Thereafter, the plate was mixed on an orbital shaker at 250 rpm (Heidolph Polymax Orbital, Labotec, 1040) for 2 min to induce lysis and incubated for 10 min at room temperature in the dark. This resulted into the production of a luminescent signal that was measured using a Multilabel Counter (Perkin Elmer, VICTOR3™, 1420) in relative light units (RLU). The value of the background (50 μL of media) was deducted from all experimental sample wells.
4. Results and discussion
4.1 Scanning electron microscopy (SEM)
The surface morphology of CaCO3 (Fig. 1A), CD–CaCO3 (Fig. 1B), β-CD–CaCO3 + 5-Fu (Fig. 2A and B) and γ-CD–CaCO3 + Na-L-thy (Fig. 2C and D) was examined using SEM. The average size of bare CaCO3 microparticles was found to be around 3 to 5 μm obtained by measuring 100 randomly selected microparticles. Microparticles obtained were found to be spherical porous structures. The cross-sectional SEM images of broken microparticles revealed porous channel-like structures obtained due to aggregation of CaCO3 nanoparticles during the process of co-precipitation. The average size of CD–CaCO3 microparticles was 4 to 6 μm again by measuring 100 randomly selected microparticles. Interestingly, CD–CaCO3 microparticles were found to be more porous than CaCO3 microparticles and the reason for this could be hybridization of CDs to CaCO3 microparticles during the process. | ||
| Fig. 1 (A) and (B) shows low and high magnification SEM images of bare CaCO3 particles and (D) and (E) CD–CaCO3 hybrid microparticles and (C) and (F) shows size distribution of CaCO3 and CD–CaCO3 microparticles. | ||
 | ||
| Fig. 2 (A) and (B) show low and high magnification SEM images of 5-Fu encapsulated β-CD–CaCO3 hybrid microparticles, (C) and (D) show low and high magnification SEM images of Na-L-thy encapsulated γ-CD–CaCO3 hybrid microparticles. | ||
4.2 Raman spectroscopy
Raman spectra of bare CaCO3 showed a sharp band in between 1050 and 1100 cm−1 that is a characteristic of vaterite particles as shown in Fig. 3A.12 The same characteristic vaterite band is also seen in the CD–CaCO3 (Fig. 3B), 5-Fu loaded CD–CO3 (Fig. 3C) and Na-L-thy loaded CD–CaCO3 (Fig. 3D) microparticles. Hence, Raman spectra of the CD–CaCO3 microparticles before and after drug loading are identical, corresponding to the presence of vaterite in the microparticles. | ||
| Fig. 3 Raman spectra of bare (A) CaCO3 microparticles, (B) CD–CaCO3 hybrid microparticles, (C) β-CD–CaCO3 + 5-Fu and (D) γ-CD–CaCO3 + Na-L-thy. | ||
4.3 Thermogravimetric analysis
Thermogravimetric analysis (TGA) was used to find the amount of β-CD present in the CaCO3 hybrid particles. Fig. 4 shows a typical TGA analysis of the CaCO3 microparticles, prepared in the presence of β-CDs (red colour plot) (CD–CaCO3) versus those with CaCO3 only (black colour plot). An additional weight loss step is seen in the CD–CaCO3 particles (around 320 °C) compared to pure CaCO3, which is due to desorption or decomposition of β-CDs present in the matrix of CD–CaCO3 particles, compared to pure β-CDs at higher temperature can be attributed to strong interaction between the calcium carbonate nanoparticles and β-CDs in hybrid particles. The hybrid particles contain approximately ∼1.0% (by weight) β-CD in the porous matrix of hybrid CD–CaCO3 microparticles as shown in inset in Fig. 4. | ||
| Fig. 4 TGA analysis of bare CaCO3 (black colour), CD–CaCO3 microparticles (red colour) and change in the weight between CD–CaCO3 and bare CaCO3 is shown as a percentage of the total weight in inset. | ||
4.4 Brunauer–Emmett–Teller (BET) analysis
The specific area, pore size and pore volume distributions of the CaCO3 and CD–CaCO3 microparticles were investigated using Brunauer–Emmett–Teller analysis under standard temperature and pressure (Table 1). The BET surface area of CD–CaCO3 was 4.5 m2 g−1, which is slightly higher than that of the CaCO3 microparticles (4.4 m2 g−1). Moreover the pore size of CD–CaCO3 also shows a slightly wider distribution of about 441 Å, which is larger than that of the CaCO3 microparticles (421 Å). In either case, porosity is important in the delivery of hydrophobic drugs as it affects the rate of particle disintegration and payload release.| Parameter | CaCO3 | CD–CaCO3 |
|---|---|---|
| BET surface | 4.4 m2 g−1 | 4.5 m2 g−1 |
| Pore volume | 0.046 cm3 g−1 | 0.049 cm3 g−1 |
| Pore size | 421 Å | 441 Å |
4.5 Release studies
CaCO3 porous (vaterite) particles are pH-sensitive as mentioned earlier in our study.31,39 The particles are soluble in acidic media below pH 5 due to the faster dissolution of the CaCO3 microparticles but they are not soluble at neutral pH.11 The transformation of stable calcite crystals from spherical vaterite particles in aqueous medium is associated with decrease in particle size.6 This favours their use as pH responsive drug delivery systems. We therefore studied the release profile of 5-Fu loaded β-CD–CaCO3 microparticles at pH 4.8 (similar to the acidic pH in many tumour tissues) and pH 7.4 (physiologically neutral pH). Under “normal” neutral cellular conditions, less than 15% of the 5-Fu is released even after 120 h whereas about 40% release is released at pH 4.8 during the same time (Fig. 5A).6 Thus it suggests that the drug delivery system should have smaller toxicity at pH 7.4 for normal tissue cells as compared to pH 4.8. In the case of Na-L-thyroxine loaded γ-CD–CaCO3 microparticles, the release profiles were studied at pH 1.2 (acidic pH in stomach) and pH 7.4 (neutral pH), and the results are shown in Fig. 5B. At pH 1.2, the entire drug was released within 12 h, whereas at neutral pH only 30% was released in double the time, this is due to quick recrystallization of γ-CD–CaCO3 porous vaterite particles into calcite crystals in high acidic pH. In the acidic pH medium, the porous γ-CD–CaCO3 particles dissolved and recrystallizes slowly into its most stable form calcite or rhombohedral form as shown in Fig. 6A and C.40 During the recrystallization process, the entrapped CDs and drug molecules are released out. But these CaCO3 particles are stable in the basic or neutral pH (Fig. 6B and D).11,41 This pH-dependent release of hydrophobic drugs from porous CD–CaCO3 particles makes these particles potential drug carriers for pH triggered release of hydrophobic drugs in localized acidic areas as in a solid tumour and intracellular endosome as explained schematically in Fig. 7.20 | ||
| Fig. 5 (A) Shows the release profiles of 5-Fu loaded β-CD–CaCO3 particles at pH 7.4 (red colour) and at pH 4.8 (black colour). (B) Shows release profiles of Na-L-thyroxine loaded γ-CD–CaCO3 particles at pH 7.4 (red colour) and at pH 1.2 (black colour) (values present mean of three measurements ± standard error). | ||
 | ||
| Fig. 6 (A) and (B) show the SEM images of the 5-Fu loaded β-CD–CaCO3 particles after drug release for 120 h at pH 4.8 and 7.4 respectively; (C) and (D) shows Na-L-thy loaded γ-CD–CaCO3 particles after drug release for 24 h at pH 1.2 and 7.4 respectively. | ||
 | ||
| Fig. 7 Schematic representation of the release mechanism for 5-Fu loaded particles (β-CD–CaCO3 + 5-Fu) (A) where carriers are in a pure vaterite phase loaded with 5-Fu at pH 7.4 (B) whereas recrystallization in the cancerous cells is observed at slightly acidic pH 4.8 (C). | ||
4.6 Mathematical modelling
Drug release data was modelled against a zero order, first order, Higuchi model, Hixson–Crowell and Korsemeyer–Peppas model for β-CD–CaCO3 + 5-Fu and γ-CD–CaCO3 + Na-L-thy (Tables 2 and 3).| pH | Zero order (r2) | First order (r2) | Higuchi (r2) | Korsemeyer–Peppas (r2) | Hixson–Crowell (r2) |
|---|---|---|---|---|---|
| 4.8 | 0.875 | 0.676 | 0.999 | 0.752 | 0.994 |
| 7.4 | 0.971 | 0.954 | 0.971 | 0.902 | 0.975 |
| pH | Zero order (r2) | First order (r2) | Higuchi (r2) | Korsemeyer–Peppas (r2) | Hixson–Crowell (r2) |
|---|---|---|---|---|---|
| 1.2 | 0.947 | 0.765 | 0.986 | 0.850 | 0.986 |
| 7.4 | 0.815 | 0.498 | 0.932 | 0.619 | 0.920 |
Eqn (1) is based on the zero order kinetics, indicating uniform drug release from the matrix with respect to the time. It is represented by the following equation:
Zero order kinetics: describes release from porous matrices.42
 | (1) |
 is the fractional drug release, K0 is the zero-order kinetic constant and t is the time.
is the fractional drug release, K0 is the zero-order kinetic constant and t is the time.
First order kinetics: describes systems for which the rate of the drug release is dependent on the drug concentration. It is represented by following equation:
 | (2) |
Higuchi model44 describes drug release from a matrix as diffusion process based on Fick's law which represented as the square root of a time dependent process.45 The equation for the Higuchi model is as follows:
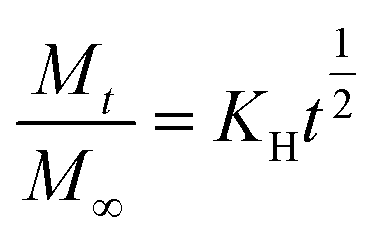 | (3) |
 is the fraction drug release, and KH is the Higuchi constant.
is the fraction drug release, and KH is the Higuchi constant.
Hixson–Crowell cube root model: is based on the mechanism of drug release depends upon the change in surface area or particle size.46
| M01/3 − Mt1/3 = KHCt | (4) |
Korsemeyer–Peppas model: represents power law release kinetics which follows non-Fickian release mechanism used to describe drug release that has an exponential relationship with respect to time.
 | (5) |
For the 5-Fu loaded microparticles (β-CD–CaCO3 + 5-Fu), the correlation coefficient (r) values of the Higuchi model was found to be higher than other models r2 = 0.999 and 0.9716 at pH 4.8 and 7.4 respectively. Similarly, for Na-L-thy loaded microparticles (γ-CD–CaCO3), the Higuchi model showed the best fit with r2 = 0.986 and 0.932 at pH 1.2 and 7.4 respectively. Thus the fraction of the drug released is proportional to the square root of time.
For CD–CaCO3 microparticles, the release rate is higher in acidic pH (1.2 or 4.8) than in phosphate buffer solution (7.4), this phenomenon is being explained by higher swelling degree of polymer networks at pH 1.2 or 4.8 compared to that at pH 7.4, due to the presence of the cyclodextrin in the microparticle structures. The hydrophilicity of CDs acts as a surfactant which helps to lower interfacial tension between the drug and the hydrophobic cavity enhancing the wettability of drug by forming inclusion complex.48 CDs serve as molecular valves to switch the ON/OFF release of payload from microparticle hybrid system.
4.7 Cell internalization
The number of internalized particles per cell was evaluated after 24 h incubation by counting the particles inside A549 cells via fluorescence microscopy and subsequent image analysis. The live cells were immediately analyzed by confocal imaging, using a multiphoton microscope. The A549 cells were stained with a green Cell Tracker (FITC) prior to the imaging. The cells were fluorescently stained and the co-localization of the fluorescently labelled CD–CaCO3 microparticles within the cytoplasm was evaluated with z-stack images as depicted in Fig. 8. | ||
| Fig. 8 Orthogonal view of β-CD–CaCO3 microparticles from different planes (x/y, x/z or y/z) of the confocal microscope images used to analyze the particle uptake. Co-localization of fluorescently labelled β-CD–CaCO3 microparticles were FITC labelled. Scale bars correspond to 20 μm. | ||
4.8 Cellular viability
The ability of cells to take up the trypan blue dye is used as an indication of cell damage. As shown in Fig. 9, when compared to untreated A549 cells, cells treated with 100 μM mL−1 of 5-Fu and those treated under all three concentrations of β-CD–CaCO3 + 5-Fu led to a significant decrease in cell viability. This decreased was concentration dependent with estimated an IC50 of 200 μM mL−1. 5-Fu has been reported to induce apoptosis and cell death.49 The combination of MTX and 5Fu, was previously reported to be more effective therapy against gastric cancer cells.50 In our current study, the combination of 5-Fu and CD–CaCO3 led to improved therapeutic effects of the 5-Fu in lung cancer cells. | ||
| Fig. 9 Trypan blue percentage viability of A549 cells on β-CD–CaCO3, 5-Fu and β-CD–CaCO3 + 5-Fu with different concentrations (25, 50 and 100 μM mL−1). Decrease in cell viability was observed with the β-CD–CaCO3 + 5-Fu when compared to 5-Fu or untreated control and significant differences (p < 0.05*) (p < 0.01**) were noted | ||
4.9 Cell proliferation
In the present study, the rate of proliferation of A549 cells after treatment with different formulations is studied (Fig. 10). Treated cells with 5-Fu and β-CD–CaCO3 + 5-Fu resulted in a decreased proliferation rate when compared to untreated control cells. In another study, 5-Fu significantly inhibited both cell proliferation and DNA synthesis in keloid fibroblasts, which later led to significant cell apoptosis.51 5-Fu at a concentration of 100 μM mL−1 was able to induce a significant decrease in cell proliferation. Upon its conjunction with CD–CaCO3, a greater decrease in cell proliferation was detected when compared to the decreased seen when 5-Fu alone was used. Cell death is the ultimate process before the cell ceases to perform its function. For cell death to occur both structure damage as well as decrease (cessation) activity (caused by decrease ATP) has to happen before cell death. The structure further determines its ability to perform any function. The combination has to have a better therapeutic effect in lung cancer cells. | ||
| Fig. 10 ATP luminescence assay of A549 cells on β-CD–CaCO3, 5-Fu and β-CD–CaCO3 + 5-Fu with different concentration (25, 50 and 100 μM mL−1). Decrease in ATP levels was observed with most of β-CD–CaCO3 + 5-Fu treated cells when compared to the untreated control and significant differences (p < 0.05*) (p < 0.01**) (p < 0.001***) were noted. | ||
4.10 Cell cytotoxicity
The CytoTox96® nonradioactive cytotoxicity assay was performed on cells treated with β-CD–CaCO3, 5-Fu, or β-CD–CaCO3 + 5-Fu at different concentration as shown in Fig. 11. All the concentrations of 5-Fu and CDCO3 + 5-Fu led to significant increase in the level of LDH and cytotoxicity. The anticancer drug, 5-Fu is well known for its cytotoxic effects and nuclear damage. The study confirmed the ability of 5-Fu to induce damages in cancer cells. In another study, the potent cytotoxic effects of 5-fluorouracil (5-Fu) on human HeLa (uterine cervical cancer), MCF-7 (mammary cancer), WI-38 CT-1 (embryonic lung fibroblasts), KMM-1 (myeloma) and Raji (Burkitt's lymphoma) were already demonstrated.52 The current results indicated a possibility that combined therapy of certain types of anticancer drugs with CDCO3 may be effective in treatment of cancer patients.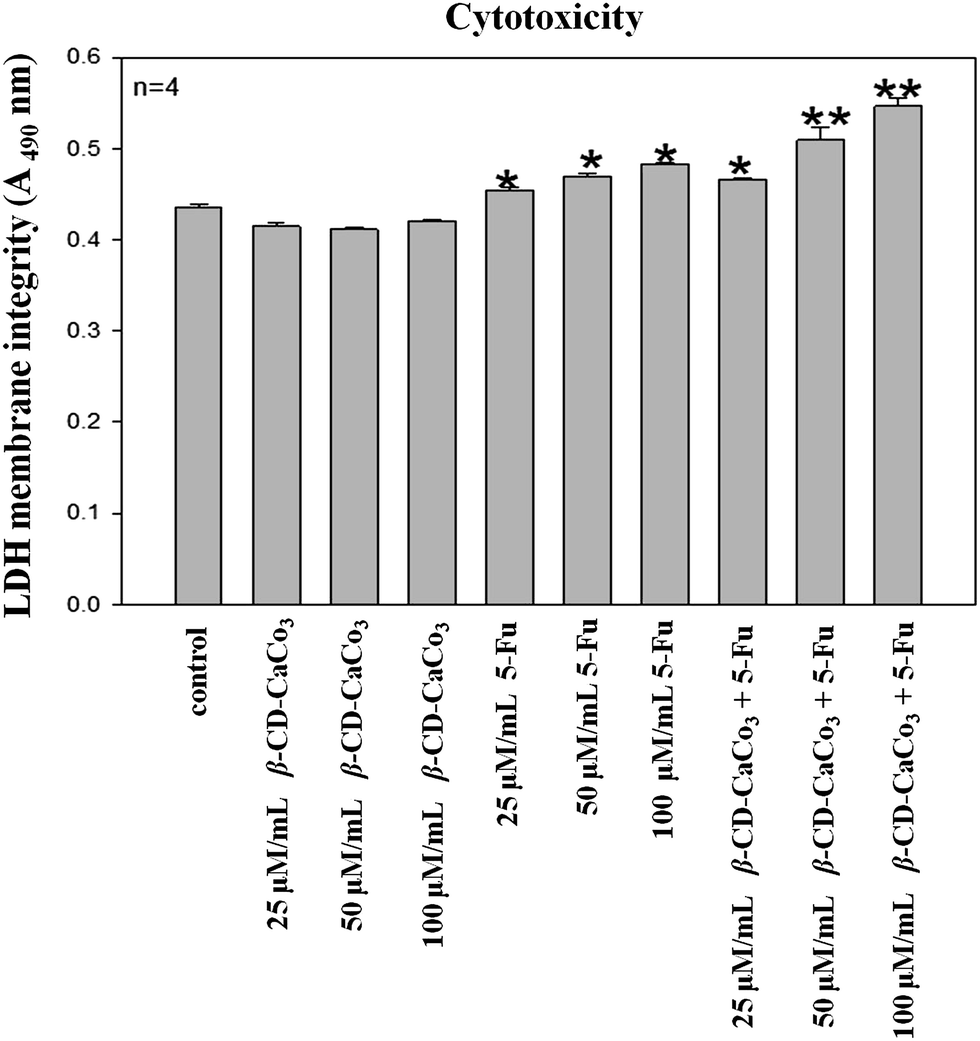 | ||
| Fig. 11 LDH cytotoxicity assay after A549 cells were subjected to various treatments. At a concentration of 100 μM mL−1 β-CD–CaCO3 + 5-Fu led to significant increase in cytotoxicity when compared to untreated control. | ||
5. Conclusions
In the present paper, a new approach to the encapsulation of an anticancer drug or hormone with polymeric microparticles has been described. It utilizes the ability of CD–CO3 microparticles, formed by the CD in calcium chloride and sodium carbonate solution to formation inclusion complex of hydrophobic drugs by entrapped CD within the porous microparticles. The suggested method makes it possible to circumvent some problems which are associated with hydrophobic drugs as compared to other available encapsulation drug delivery systems.Acknowledgements
The authors would like to acknowledge UJ-COMMONWEALTH bursary (University of Johannesburg) and Sandiswa Imbewu (Rhodes University) for funding PhD project.References
- K. Nam, H. Y. Nam, P.-H. Kim and S. W. Kim, Biomaterials, 2012, 33, 8122–8130 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2012, 54, 631–651 CrossRef.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- M. Arruebo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 16–30 CrossRef CAS PubMed.
- C. Peng, Q. Zhao and C. Gao, Colloids Surf., A, 2010, 353, 132–139 CrossRef CAS.
- D. V. Volodkin, R. von Klitzing and H. Möhwald, Angew. Chem., Int. Ed., 2010, 49, 9258–9261 CrossRef CAS PubMed.
- K. Gopal, Z. Lu, M. M. de Villiers and Y. Lvov, J. Phys. Chem. B, 2006, 110, 2471–3247 CrossRef CAS PubMed.
- X. Li, Q. Hu, L. Yue and J. Shen, Chem.–Eur. J., 2006, 12, 5770–5778 CrossRef CAS PubMed.
- W. Wei, G.-H. Ma, G. Hu, D. Yu, T. Mcleish, Z.-G. Su and Z.-Y. Shen, J. Am. Chem. Soc., 2008, 130, 15808–15810 CrossRef CAS PubMed.
- B. V. Parakhonskiy, A. Haase and R. Antolini, Angew. Chem., Int. Ed., 2012, 51, 1195–1197 CrossRef CAS.
- D. V. Volodkin, A. I. Petrov, M. Prevot and G. B. Sukhorukov, Langmuir, 2004, 20, 3398–3406 CrossRef CAS PubMed.
- S. Schmidt and D. Volodkin, J. Mater. Chem. B, 2013, 1, 1210–1218 RSC.
- A. Natoli, M. Wiens, H.-C. Schröder, M. Stifanic, R. Batel, A. L. Soldati, D. E. Jacob and W. E. Müller, Micron, 2010, 41, 359–366 CrossRef CAS PubMed.
- N. G. Balabushevich, A. V. Lopez de Guerenu, N. A. Feoktistova and D. Volodkin, Phys. Chem. Chem. Phys., 2015, 201, 2523 RSC.
- K. Naka, Y. Tanaka and Y. Chujo, Langmuir, 2002, 18, 3655–3658 CrossRef CAS.
- A. López-Macipe, J. Gómez-Morales and R. Rodriguez-Clemente, J. Cryst. Growth, 1996, 166, 1015–1101 CrossRef.
- M. Yang, X. Jin and Q. Huang, Colloids Surf., A, 2011, 374, 102–107 CrossRef CAS.
- Y. Svenskaya, B. Parakhonskiy, A. Haase, V. Atkin, E. Lukyanets, D. Gorin and R. Antolini, Biophys. Chem., 2013, 182, 11–15 CrossRef CAS PubMed.
- N. Qiu, H. Yin, B. Ji, N. Klauke, A. Glidle, Y. Zhang, H. Song, L. Cai, L. Ma and G. Wang, Mater. Sci. Eng., C, 2012, 32, 2634–2640 CrossRef CAS.
- F. L. Aachmann, D. Otzen, K. L. Larsen and R. Wimmer, Protein Eng., 2003, 16, 905–912 CrossRef CAS PubMed.
- E. Bilensoy, O. Gürkaynak, A. L. Doğan and A. A. Hıncal, Int. J. Pharm., 2008, 347, 163–170 CrossRef CAS PubMed.
- A. Mahmoud, G. S. El-Feky, R. Kamel and G. E. Awad, Int. J. Pharm., 2011, 413, 229–236 CrossRef CAS PubMed.
- S. Sajesh and C. P. Sharma, Int. J. Pharm., 2006, 325, 147–215 CrossRef PubMed.
- M. Cirri, M. Bragagni, N. Mennini and P. Mura, Eur. J. Pharm. Biopharm., 2012, 80, 46–53 CrossRef CAS PubMed.
- A. Trapani, A. Lopedota, M. Franco, N. Cioffi, E. Ieva, M. Garcia-Fuentes and M. J. Alonso, Eur. J. Pharm. Biopharm., 2010, 75, 26–32 CrossRef CAS PubMed.
- K. Wang, Y. Liu, C. Li, S.-X. Cheng, R.-X. Zhuo and X.-Z. Zhang, ACS Macro Lett., 2013, 2, 201–205 CrossRef CAS.
- S. S. Dhule, P. Penfornis, T. Frazier, R. Walker, J. Feldman, G. Tan, J. He, A. Alb, V. John and R. Pochampally, Nanomedicine, 2012, 8, 440–451 CAS.
- S. Sajeesh, K. Bouchemal, V. Marsaud, C. Vauthier and C. P. Sharma, J. Controlled Release, 2010, 147, 377–384 CrossRef CAS PubMed.
- H. Wang, N. Shao, S. Qiao and Y. Cheng, J. Phys. Chem. B, 2012, 116, 11217–11224 CrossRef CAS PubMed.
- R. Kurapati and A. M. Raichur, J. Mater. Chem. B, 2013, 1, 3175–3184 RSC.
- J. Marchal, H. Boulaiz, F. Rodriguez-Serrano, M. Peran, E. Carrillo, C. Velez, J. Dominguez, J. Gomez-Vidal, J. Campos and M. Gallo, Med. Chem., 2007, 3, 233–239 CrossRef CAS.
- S. Li, A. Wang, W. Jiang and Z. Guan, BMC Cancer, 2008, 8, 103 CrossRef PubMed.
- R. Aydin and M. Pulat, J. Nanomater., 2012, 2012, 1–10 CrossRef.
- S.-y. Wu, W. L. Green, W.-s. Huang, M. T. Hays and I. J. Chopra, Thyroid, 2005, 15, 943–958 CrossRef CAS PubMed.
- N. Messier, L. Laflamme, G. Hamann and M. Langlois, Mol. Cell. Endocrinol., 2001, 174, 59–69 CrossRef CAS PubMed.
- J. Lakkakula, R. W. M. Krause, D. T. Ndinteh, S. P. Vijaylakshmi and A. M. Raichur, J. Inclusion Phenom. Macrocyclic Chem., 2012, 74, 397–405 CrossRef CAS.
- G. B. Sukhorukov, D. V. Volodkin, A. M. Gunther, A. I. Petrov, D. B. Shenoy and H. Mohwald, J. Mater. Chem., 2004, 14, 2073–2081 RSC.
- N. A. Dhas and K. S. Suslick, J. Am. Chem. Soc., 2005, 127, 2368–2369 CrossRef CAS PubMed.
- N. Spanos and P. G. Koutsoukos, J. Cryst. Growth, 1998, 191, 783–790 CrossRef CAS.
- J. Gómez-Morales, J. Torrent-Burgués and R. Rodríguez-Clemente, J. Cryst. Growth, 1996, 169, 331–3388 CrossRef.
- H. M. Mansour, M. Sohn, A. Al-Ghananeem and P. P. DeLuca, Int. J. Mol. Sci., 2010, 11, 3298–3322 CrossRef CAS PubMed.
- Y. M. Amgaokar, R. V. Chikhale, U. B. Lade, D. M. Biyani and M. J. Umekar, Dig. J. Nanomater. Biostruct., 2011, 6, 475–497 Search PubMed.
- T. Higuchi, J. Pharm. Sci., 1963, 52, 1145–1149 CrossRef CAS PubMed.
- J. Siepmann and N. Peppas, Adv. Drug Delivery Rev., 2012, 48, 139–157 CrossRef.
- A. Hixson and J. Crowell, Ind. Eng. Chem., 1931, 23, 923–931 CrossRef CAS.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef CAS.
- S. S. Shah, T. Y. Pasha, A. K. Behera and A. Bhandari, Pharm. Lett., 2012, 4, 354–366 CAS.
- M. C. Filgueiras, A. Morrot, P. M. G. Soares, M. L. Costa and C. Mermelstein, PLoS One, 2013, 8, e63177 CAS.
- Y. Takekazu, S. Yasuhiro, S. Kuniaki, O. Atsushi, I. Nobumasa, H. Ichinosuke, S. Hiroshi, I. Hiroaki, T. Yasushi, T. Takao, Y. Seiichiro and Y. Shigeaki, Jpn. J. Clin. Oncol., 2004, 34, 316–322 CrossRef PubMed.
- L. Huang, Y. P. Wong, Y. J. Cai, I. Lung, C. S. Leung and A. Burd, Br. J. Dermatol., 2010, 163, 1181–1185 CrossRef CAS PubMed.
- M. Toshihiro, O. Shigeo, K. Toshinori, N. Masahiro and N. Masayoshi, Cancer Lett., 1983, 17, 239–247 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |