
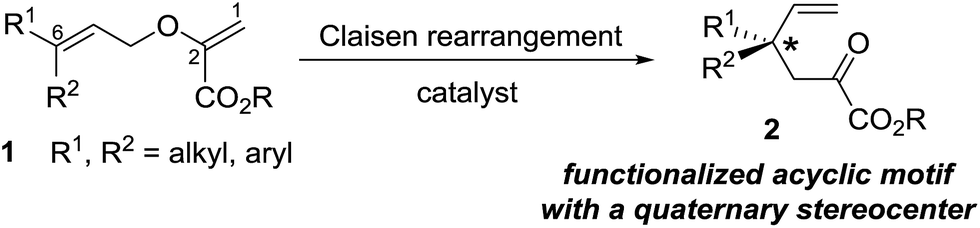
Modular synthesis of allyl vinyl ethers for the enantioselective construction of functionalized quaternary stereocenters†
Gopinathan Muthusamy and
Sunil V. Pansare*
Department of Chemistry, Memorial University, St. John's, Newfoundland, Canada A1B3X7. E-mail: spansare@mun.ca
First published on 27th October 2016
Abstract
A concise synthesis of 2-t-butoxycarbonyl allyl vinyl ethers by regioselective Petasis methylenation and stereoselective Suzuki–Miyaura cross-coupling reactions of iodoallyl t-butyl oxalates was developed. Ethers with a terminally unsubstituted vinyl group and a terminally disubstituted allyl portion are readily accessible by this method. The enantioselective Claisen rearrangement of representative allyl vinyl ethers was examined.
The Claisen rearrangement is a cornerstone of synthetic organic chemistry and it is one of the most extensively studied reactions.1 Applications of the rearrangement range from the construction of key structural elements in natural products to the stereoselective assembly of cyclic and acyclic motifs for applications in other synthetic endeavours.2 Catalysis of the Claisen rearrangement is of particular interest and catalytic asymmetric versions have also been intensely investigated.3 In recent years, allyl vinyl ethers with an alkoxycarbonyl group at the 2-position (the so-called Gosteli-Claisen substrates 1, Fig. 1), have attracted attention due to the potential for two-point interaction of these dienes with Lewis acids,3a,b and acceleration of the rearrangement by the electron withdrawing alkoxycarbonyl group.4 This structural feature in the allyl vinyl ethers 1 has therefore enabled the catalysis of their Claisen rearrangement.5
 | ||
| Fig. 1 Claisen rearrangement of 2-alkoxycarbonyl allyl vinyl ethers. | ||
Our interest in the Claisen rearrangement stems from our studies on the asymmetric synthesis of functionalized quaternary stereocenters.6 In this context, the rearrangement of allyl vinyl ethers 1 with a terminally unsubstituted vinyl portion and a terminally disubstituted allyl group would generate a functionalized, quaternary stereocenter-containing, acyclic motif 2 (Fig. 1). The enantioselective synthesis of such acyclic building blocks has attracted attention in recent years.7 Notably, while the Gosteli-Claisen rearrangement has been employed for establishing vicinal stereocenters,5 its utility in the synthesis of isolated, acyclic, functionalized quaternary stereocenters is unexplored.8 Herein, we report a modular synthesis of a variety of Gosteli-Claisen substrates 1 with a terminally unsubstituted vinyl group and preliminary studies on their enantioselective rearrangement to the functionalized, quaternary stereocenter containing, acyclic building blocks 2.
Despite the potential synthetic utility of dienes such as 1, only a few methods are available for their synthesis: (a) Mannich reaction of allyloxymalonates followed by quaternization and decarboxylative elimination,9a (b) aldol reaction of O-allyl glycolates followed by dehydration,9b (c) O-allylation of α-keto acid or ester enolates3d and (d) palladium catalyzed vinyl interchange reaction of alcohols and methyl 2-methoxy acrylate.9c Of these methods, the enolate O-allylation protocol requires an α-keto acid as the starting material and generally provides modest yields. In addition, it has not been used for esters of pyruvic acid. Importantly, the other syntheses of allyloxy acrylates9 require multiple steps for installing the vinyl group via Mannich9a or aldol reactions9b and the products are sometimes obtained as a mixture with the corresponding Claisen rearrangement product.9a The vinyl exchange reaction9c requires a large excess of the alcohol component (usually as the solvent) and, in our hands, attempts to achieve this conversion with ethyl 2-methoxy acrylate and stoichiometric amount of (E)-3-phenylbut-2-en-1-ol provided the vinyl exchange product in very low yield (13%). In addition, all of the above methods require access to a stereochemically defined allylic alcohol or a derivative as the starting material, which must be prepared separately for each allyl vinyl ether. Evidently, methodology that overcomes these limitations and provides stereocontrolled access to α-allyloxy acrylates would be useful.
Given the limitations of existing methods, we decided to explore a strategy that would provide the required 2-alkoxycarbonyl motif of the allyl vinyl ethers in a single step and would also offer access to a variety of terminally disubstituted allyl groups. We reasoned that both of these objectives could be achieved by the chemoselective functionalization of an unsymmetrical dialkyl oxalate. Specifically, we decided to examine the Petasis methylenation10 of oxalic acid diesters that were derived from t-butyl alcohol, for steric control of the regioselectivity11a of the methylenation,11 and from allylic alcohols that could potentially be modified by the introduction of alkyl or aryl groups via cross-coupling reactions. Allylic alcohols with a vinyl halide motif were selected for this purpose.
Our studies began with the synthesis of the allylic alcohols 3–6 (Scheme 1). (E)-3-Bromo-2-butenol (3) was obtained by the bromination/decarboxylative debromination of 3-methyl-2-furanone12a and (Z)-3-bromo-2-buten-1-ol (4) was obtained by hydrobromination12b of ethyl propargylate followed by reduction to the alcohol.12c (E)-3-Iodo-2-butenol (5) was prepared in one step12d from but-2-yn-1-ol and (Z)-3-iodo-2-buten-1-ol (6) was prepared as described for 4. Esterification of 3–6 with t-butyl oxalyl chloride12e provided the unsymmetrical dialkyloxalates 7–10 (77–89%, Scheme 1). Pleasingly, the Petasis methylenation of 7–10, by heating with dimethyl titanocene under microwave irradiation, provided the corresponding allyl vinyl ethers 11–14 (40–61%) respectively. Notably, the products of methylenation of the more hindered ester functionality were not detected in the crude reaction mixtures.13
 | ||
| Scheme 1 Petasis methylenation of haloallyl t-butyl oxalates. | ||
The cross-coupling reactions14a of 11–14 were investigated next. Initially, we explored the cross coupling reaction of the bromo derivatives 11 and 12 with a selection of aryl as well as alkyl boronic acids or their derivatives with PdCl2(dppf)·CH2Cl2 as the catalyst. After a brief survey of solvents and reaction conditions it was evident that these cross-coupling reactions either failed or provided very low yields of the required products. Hence, further studies were done with the iodo derivatives 13 and 14. Since the introduction of an alkyl group in 11/12 was particularly challenging, initial studies with 13/14 focused on their cross-coupling reactions with butyl boronic acid derivatives. Although alkyl trifluoroborates are sometimes better cross-coupling reagents than alkyl boronic acids,6a,14b the attempted cross-coupling of BuBF3K with either 13 or 14 in the presence of PdCl2(dppf)·CH2Cl2 and Cs2CO3 in toluene/H2O led to complete decomposition of the vinyl halide. The effect of Ag2O as an additive14c,d was examined next. In these studies, butyl boronic acid performed better than BuBF3K. Thus, while no cross-coupling was observed with 14 and BuBF3K the use of BuB(OH)2 provided 16a (16%) when PdCl2(dppf)·CH2Cl2 and Ag2O were used in THF, and NaHCO3 was employed as the base. The yield of 16a improved significantly (67%) when PdCl2(dppf)·CH2Cl2 was replaced with Pd(PPh3)4 and K2CO3 was used as the base.14e Using these conditions, 13 provided 15a (63%). Similarly, the cross-coupling of 13 with isobutyl- and cyclopropylboronic acids provided 15b (61%) and 15c (65%) respectively. Although the use of Ag2O as an additive was beneficial for cross-coupling reactions of 13 with aryl boronic acids, some modification of the conditions used for the cross-coupling with alkylboronic acids was also necessary. Thus, while the unoptimized cross-coupling of 13 and 2-naphthyl boronic acid (PdCl2(dppf)·CH2Cl2, Cs2CO3 in CH3CN) provided 15d in low yield (22%) in initial studies, the use of Ag2O as an additive and KOH as the base in dioxane14f considerably improved the yield of 15d (68%). With the optimized reaction conditions in hand, several alkyl as well as aryl boronic acids were coupled with 13 and 14 to provide the functionalized allyl vinyl ethers 15a–j and 16a–h and these results are summarized in Fig. 2.
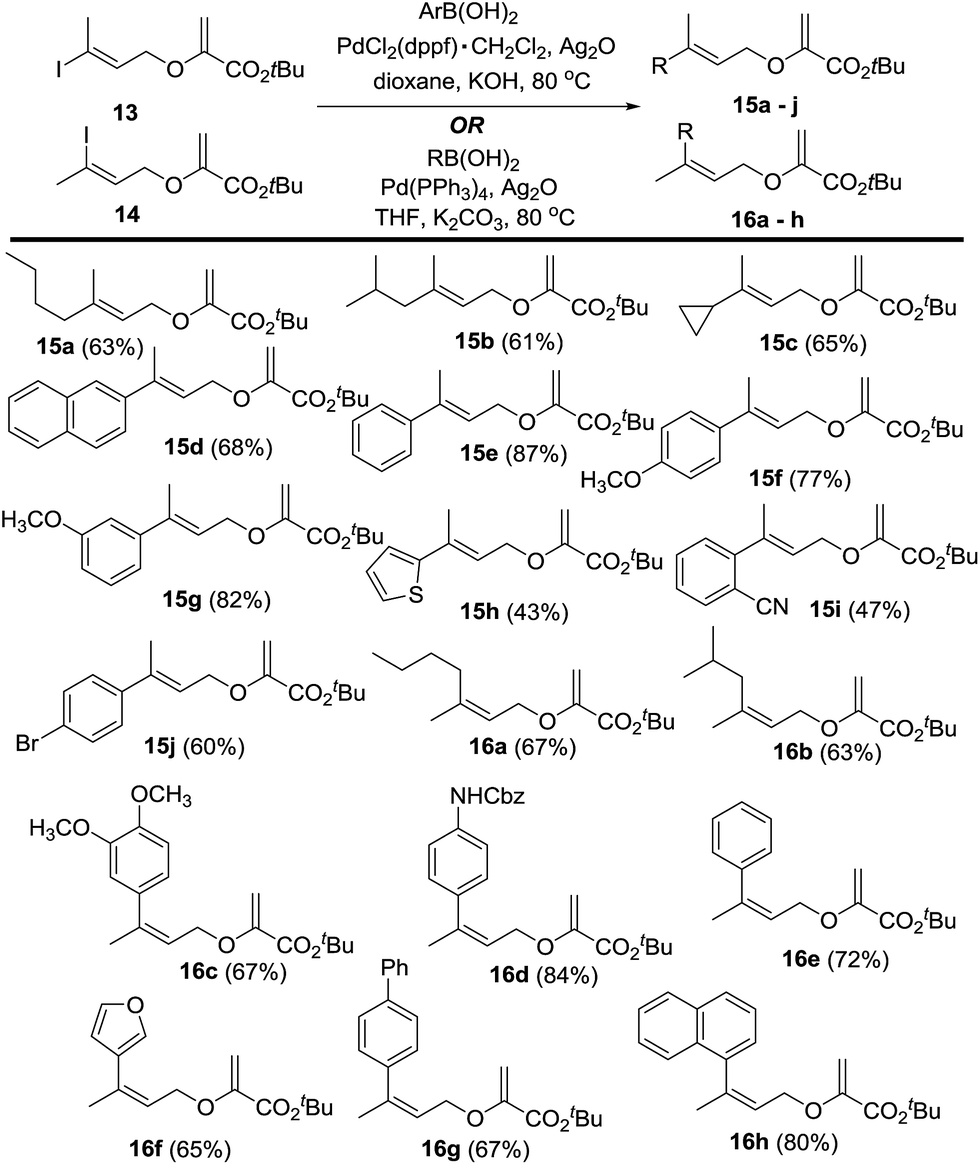 | ||
| Fig. 2 Synthesis of allyl vinyl ethers 15 and 16. | ||
Notably, although both Pd(0)15a,b and Pd(II)15c–f derived catalysts are reported to catalyze the Claisen rearrangement of allyl vinyl ethers at ambient temperature, rearrangement of 15 and 16 was not observed under the optimized reaction conditions. However, it is also known that the Pd(II) catalyzed Claisen rearrangement reactions are sensitive to the nature of the ligand in the Pd catalyst.15d The stability of 15 and 16 in the present study, despite the presence of transient Pd(II) species, underscores the importance of the ligand effect and may be attributed to the use of a diphosphine ligand on the palladium. Although this effect has not been investigated for the Claisen rearrangements with Pd(0)-derived catalytic species, the results of the present study suggest that either the nature of the Pd(0) species or the electronic properties of the substrate are critical for the rearrangement. Alternatively, it is plausible that the cross-coupling reactions of 13 and 14 are faster than the Pd-catalyzed Claisen rearrangement of 15 and 16.
Having established a synthetic protocol for the required allyl vinyl ethers 15 and 16, the possibility of an enantioselective Claisen rearrangement was examined next. In preliminary studies, exposure of several of the ethers to a selection of metal-derived catalysts resulted in facile Claisen rearrangement at ambient temperature. Promising results were obtained with the catalyst derived from Cu(OTf)2 and (R,R)-diphenylbox (L, Scheme 2)16a when diethyl ether was used as the solvent. Thus, the ethers 15a, 15e and 15j provided the α-keto esters 17a (58% yield, 98% ee), 17e (53%, 45% ee) and 17j (48%, 56% ee) respectively. The results suggest that dialkyl substitution at C-6 in the allyl vinyl ether is beneficial for enantioselectivity.
 | ||
| Scheme 2 Enantioselective Claisen rearrangement of 15 to 17. | ||
The formation of S-17 as the major enantiomer is based on a transition state assembly for related rearrangements proposed by Hiersemann.16b The allyl vinyl ether forms a bidentate chelate with the Cu(II)–bis(oxazoline) complex to generate the sterically favoured distorted square planar complex A (Scheme 2) in which one face (Re for 15a and Si for 15e and 15j) of the terminal carbon (C6) in the allyl group is exposed to the vinyl group (C1) in a chair-like conformation of the diene. The complex B is disfavoured due to steric interactions between the R substituent in the allyl portion and the phenyl ring in the bisoxazoline ligand. Subsequent rearrangement from complex A would generate the S enantiomer of 17.
The precise reasons for the lower enantioselectivity of the rearrangement of 15e and 15j are not known at this time. It is plausible that, for these substrates, the difference in energy of the complexes A and B is less than the corresponding difference in energy for the complexes of 15a, presumably due to the greater steric requirements of a butyl group in 15a compared to a phenyl group in 15e and 15j. Alternatively, resonance stabilization of positive charge at C6 (benzylic carbocation) may favour a more polar transition state (more C4–O bond cleavage than C1–C6 bond formation)17 for the rearrangement of 15e and 15j compared to 15a. This could facilitate rotation of the C5–C6 bond and expose the Re face of C6 in 15e and 15j to the vinyl group, thereby generating more of the R enantiomer of 17e and 17j.
Conclusions
In conclusion, we have developed a modular synthesis of 2-t-butoxycarbonyl allyl vinyl ethers that are unsubstituted at C1 (vinyl portion) and disubstituted at C6 (allyl portion). The methodology offers a one-step construction of the functionalized vinyl portion of the ether and enables stereospecific synthesis of the terminally disubstituted allyl functionality. Enantioselective Claisen rearrangement of the allyl vinyl ethers generates a functionalized, acyclic motif with a quaternary stereocenter. Current efforts focus on optimizing the enantioselectivity of the Claisen rearrangement and further transformations of 17.Acknowledgements
In memory of Mukund G. Kulkarni, for his contributions on the Claisen rearrangement. These investigations were supported by the Natural Sciences and Engineering Research Council of Canada and the Canada Foundation for Innovation.Notes and references
- A. M. Castro and M. Tortosa, in Comprehensive Organic Synthesis, ed. P. Knochel and G. A. Molander, 2nd edn, 2014, vol. 5, p. 912 Search PubMed.
- R. A. Fernandes, A. K. Chowdhury and P. Kattanguru, Eur. J. Org. Chem., 2014, 2833 CrossRef CAS.
- (a) J. Rehbein and M. Hiersemann, Synthesis, 2013, 45, 1121 CrossRef CAS; (b) M. Hiersemann and L. Abraham, Eur. J. Org. Chem., 2002, 1461 CrossRef CAS; (c) L. Abraham, R. Czerwonka and M. Hiersemann, Angew. Chem., Int. Ed., 2001, 40, 4700 CrossRef CAS; (d) C. Uyeda and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 9228 CrossRef CAS PubMed; (e) T. C. A. F. Rodrigues, W. A. Silva and A. H. L. Machado, Curr. Org. Synth., 2015, 12, 795 CrossRef CAS.
- Rate enhancement of Claisen rearrangement by the trifluoromethyl and the cyano group at C-2 in the allyl vinyl ether is also known, see: (a) J. J. Gajewski, K. R. Gee and J. Jurayj, J. Org. Chem., 1990, 55, 1813 CrossRef CAS; (b) C. J. Burrows and B. K. Carpenter, J. Am. Chem. Soc., 1981, 103, 6983 CrossRef CAS; (c) V. Aviyente, H. Y. Yoo and K. N. Houk, J. Org. Chem., 1997, 62, 6121 CrossRef CAS; (d) For an informative summary of the effect of substituents on the rate of the Claisen rearrangement, see: N. F. O'Rourke and J. E. Wulff, Org. Biomol. Chem., 2014, 12, 1292 RSC.
- (a) J. Rehbein and M. Hiersemann, in Asymmetric Synthesis (II), ed. M. Christmann and S. Bräse, 2012, p. 157 Search PubMed and references therein; (b) J. Troendlin, J. Rehbein, M. Hiersemann and O. Trapp, J. Am. Chem. Soc., 2011, 133, 16444 CrossRef CAS PubMed; (c) T. Ollevier and T. M. Mwene-Mbeja, Can. J. Chem., 2008, 86, 209 CrossRef CAS; (d) C. Grison, T. K. Olszewski, C. Crauste, A. Fruchier, C. Didierjean and P. Coutrot, Tetrahedron Lett., 2006, 47, 6583 CrossRef CAS.
- (a) A. Manchoju, R. G. Thorat and S. V. Pansare, Eur. J. Org. Chem., 2015, 5939 CrossRef CAS; (b) S. V. Pansare and A. Bhattacharyya, Tetrahedron Lett., 2001, 42, 9265 CrossRef CAS.
- (a) C. Park, M. W. Ha, B. Kim, S. Hong, D. Kim, Y. Park, M. Kim, J. K. Lee, J. Lee and H. Park, Adv. Synth. Catal., 2015, 357, 2841 CrossRef CAS; (b) Y. Shi, B. Jung, S. Torker and A. H. Hoveyda, J. Am. Chem. Soc., 2015, 137, 8948 CrossRef CAS PubMed; (c) S. Chen, Q. Lou, Y. Ding, S. Zhang, W. Hu and J. Zhao, Adv. Synth. Catal., 2015, 357, 2437 CrossRef CAS; (d) T. Tatsumi, T. Misaki and T. Sugimura, Chem. Eur. J., 2015, 21, 18971 CrossRef CAS PubMed; (e) B. W. H. Turnbull and P. A. Evans, J. Am. Chem. Soc., 2015, 137, 6156 CrossRef CAS PubMed; (f) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593 RSC; (g) Y. Minko and I. Marek, Chem. Commun., 2014, 50, 12597 RSC; (h) M. Fañanás-Mastral, R. Vitale, M. Pérez and B. L. Feringa, Chem. Eur. J., 2015, 21, 4209 CrossRef PubMed; (i) Y. Liu, X. Liu, H. Hu, J. Guo, Y. Xia, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 4054 CrossRef CAS PubMed.
- To the best of our knowledge, only a sole example of such a stereoselective rearrangement has been described, see ref. 3a.
- (a) J. J. Gajewski, J. Jurayj, D. R. Kimbrough, M. E. Gande, B. Ganem and B. K. Carpenter, J. Am. Chem. Soc., 1987, 109, 1170 CrossRef CAS; (b) M. Hiersemann, Synthesis, 2000, 1279 CrossRef CAS; (c) G. A. Divers and G. A. Berchtold, Synth. Commun., 1977, 7, 43 CrossRef CAS.
- N. A. Petasis and E. I. Bzowej, J. Am. Chem. Soc., 1990, 112, 6392 CrossRef CAS . For a review on alkylidenation of carboxylic acid derivatives, see: R. C. Hartley and G. J. McKiernan, J. Chem. Soc., Perkin Trans. 1, 2002, 2763 RSC.
- (a) M. J. Cook, D. W. Fleming and T. Gallagher, Tetrahedron Lett., 2005, 46, 297 CrossRef CAS; (b) H. K. Chenault and L. F. Chafin, J. Org. Chem., 1994, 59, 6167 CrossRef CAS; (c) E. Vedejs and S. M. Duncan, J. Org. Chem., 2000, 65, 6073 CrossRef CAS PubMed.
- (a) C.-G. Cho, W.-S. Kim and A. B. Smith III, Org. Lett., 2005, 7, 3569 CrossRef CAS PubMed; (b) X.-Y. Lu, G.-X. Zhu and S.-M. Ma, Chin. J. Chem., 1993, 11, 267 CrossRef CAS; (c) E. Piers, C. L. Harrison and C. Zetina-Rocha, Org. Lett., 2001, 3, 3245 CrossRef CAS PubMed. The ester was reduced to the alcohol 4 with LAH instead of DIBAL as described in this report; (d) F. E. McDonald, K. Ishida and J. A. Hurtak, Tetrahedron, 2013, 69, 7746 CrossRef CAS. The yield of 5 was higher (70%) than the reported yield (31%); (e) B. Zhao and T.-P. Loh, Org. Lett., 2013, 15, 2914 CrossRef CAS PubMed.
- The ethyl ester analogs of 7 and 8, prepared from ethyl oxalyl chloride, provided an inseparable mixture of mono-methylenation products (enol ethers) that were obtained by reaction of either the ethyl ester or the allyl ester portion of the oxalate.
- (a) Review on cross-coupling reactions of alkyl boronic acids: H. Doucet, Eur. J. Org. Chem., 2008, 2013 CrossRef CAS; (b) G. A. Molander and N. Ellis, Acc. Chem. Res., 2007, 40, 275 CrossRef CAS PubMed; (c) T. Gillmann and T. Weeber, Synlett, 1994, 649 CrossRef CAS; (d) Review on Ag-mediated coupling reactions: J.-M. Weibel, A. Blanc and P. Pale, Chem. Rev., 2008, 108, 3149 CrossRef CAS PubMed; (e) G. Zou, Y. K. Reddy and J. R. Falck, Tetrahedron Lett., 2001, 42, 7213 CrossRef CAS; (f) H. Chen and M.-Z. Deng, J. Org. Chem., 2000, 65, 4444 CrossRef CAS PubMed.
- (a) U. Kazmaier, J. Org. Chem., 1994, 59, 6667 CrossRef CAS; (b) P. Koukal, H. Dvořáková, D. Dvořák and T. Tobrman, Chem. Pap., 2013, 67, 3 CAS; (c) M. Sugiura, M. Yanagisawa and T. Nakai, Synlett, 1995, 447 CrossRef CAS; (d) K. Akiyama and K. Mikami, Tetrahedron Lett., 2004, 45, 7217 CrossRef CAS; (e) N. J. Kerrigan, C. J. Bungard and S. G. Nelson, Tetrahedron, 2008, 64, 6863 CrossRef CAS; (f) T. Cao, E. C. Linton, J. Deotch, S. Berritt and M. C. Kozlowski, J. Org. Chem., 2012, 77, 11034 CrossRef CAS PubMed.
- For a review on chiral bis(oxazoline)–copper(II) complexes, see: (a) J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325 CrossRef CAS PubMed; (b) L. Abraham, M. Köerner and M. Hiersemann, Tetrahedron Lett., 2004, 12, 1292 Search PubMed; (c) J. Tan, C.-H. Cheon and H. Yamamoto, Angew. Chem., Int. Ed., 2012, 51, 8264 CrossRef CAS PubMed.
- Polar transition states for the Claisen rearrangement have been proposed on the basis of computational studies as well as experimentally observed substituent effects, see: (a) H. Y. Yoo and K. N. Houk, J. Am. Chem. Soc., 1997, 119, 2877 CrossRef CAS; (b) L. Abraham, M. Körner and M. Hiersemann, Tetrahedron Lett., 2004, 45, 3647 CrossRef CAS; (c) R. M. Coates, B. D. Rogers, S. J. Hobbs, D. R. Peck and D. P. Curran, J. Am. Chem. Soc., 1987, 109, 1160 CrossRef CAS . Also see ref. 4a–d.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods and spectroscopic data for all compounds. See DOI: 10.1039/c6ra24284g |
| This journal is © The Royal Society of Chemistry 2016 |