
Synthesis and cytotoxic evaluation of new terpenylpurines†
Abstract
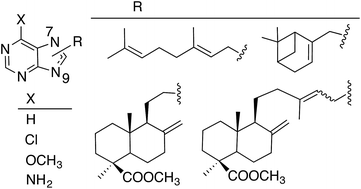
Several new terpenylpurine derivatives were prepared through alkylation of different purines with halogenated reagents derived from natural terpenoids, commercially available or isolated from cones of C. sempervirens L. and further transformed into appropriate alkylated agents. Alkylation of the purines gave mixtures of 9- and 7-alkylpurines, being the 9-alkylpurines the major regioisomers. The presence of the terpenyl residue induced cytotoxicity on simple purines and, in general, that activity improved as the substituent was larger. The 7-diterpenyl-6-chloropurine E-21b was the most cytotoxic in the series and it can be considered an analogue of the marine natural compounds agelasines and agelasimines, which were taken as models for this work.
 Please wait while we load your content...
Please wait while we load your content...

