
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of carbon substrate on the electrochemical performance of carbon/manganese oxide hybrids in aqueous and organic electrolytes†
Marco Zeigerab,
Simon Fleischmannab,
Benjamin Krünerab,
Aura Tolosaab,
Stephan Bechtelab,
Mathias Baltesb,
Anna Schreibera,
Riko Moronic,
Severin Vierrathc,
Simon Thielecd and
Volker Presser*ab
aINM - Leibniz Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany. E-mail: volker.presser@leibniz-inm.de
bDepartment of Materials Science and Engineering, Saarland University, Campus D2 2, 66123 Saarbrücken, Germany
cIMTEK Department of Microsystems Engineering, University of Freiburg, Georges-Koehler-Allee 105, 79110 Freiburg, Germany
dFIT, University of Freiburg, Georges-Koehler-Allee 103, 79110, 79104 Freiburg, Germany
First published on 25th October 2016
Abstract
Manganese oxide presents very promising electrochemical properties as an electrode material in supercapacitors, but there remain important open questions to guide further development of the complex manganese oxide/carbon/electrolyte system. Our work addresses specifically the influence of carbon ordering and the difference between outer and inner porosity of carbon particles for the application in aqueous 1 M Na2SO4 and 1 M LiClO4 in acetonitrile. Birnessite-type manganese oxide was hydrothermally hybridized on two kinds of carbon onions with only outer surface area and different electrical conductivity, and conventional activated carbon with a high inner porosity. Carbon onions with a high degree of carbon ordering, high conductivity, and high outer surface area were identified as the most promising material, yielding 179 F g−1. Pore blocking in activated carbon yields unfavorable electrochemical performances. The highest specific energy of 16.4 W h kg−1 was measured for a symmetric full-cell arrangement of manganese oxide coated high temperature carbon onions in the organic electrolyte. High stability during 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles was achieved for asymmetric full-cells, which proved as a facile way to enhance the electrochemical performance stability.
000 cycles was achieved for asymmetric full-cells, which proved as a facile way to enhance the electrochemical performance stability.
1. Introduction
Supercapacitors are fast and efficient electrochemical energy storage devices.1–4 Typically, they are classified in electrical double-layer capacitors (EDLCs), pseudocapacitors, and hybrid devices. EDLCs store energy via electrosorption of electrolyte ions at the charged surface of high surface area carbons, which is a capacitive, non-faradaic process. The physical nature of ion electrosorption in EDLCs enables faster and more efficient charge storage compared to chemically-driven redox reactions. Therefore, EDLCs are promising for high power applications utilizing rapid charge and discharge rates, moreover present high efficiency, and long lifetimes. Current EDLC devices suffer from low specific energies, compared to conventional batteries.1,2,5–7 To overcome this issue, the energy storage capacity can be increased by improving the properties of the electrode material, the electrolyte, and the cell preparation/design. General requirements for suitable electrode materials are a high specific surface area (SSA) with well-developed pore size distribution, good conductivity, and high electrochemical stability.8,9 A variety of carbon materials has been studied so far, including activated carbon (AC),10,11 carbide-derived carbons,12–14 templated carbons,15 onion-like carbon (OLC),16,17 graphene,18,19 carbon nanotubes (CNTs),20,21 carbon nanofibers,22–24 and carbon aerogels.25 AC is currently the most commonly used electrode material; typically, ACs present surface areas in the range of 1500–2000 m2 g−1 resulting in capacitances of 100–200 F g−1 and specific energies between 2 W h kg−1 and 20 W h kg−1 (depending on the electrolyte and operation parameters).1The ability to improve the specific energy of EDLCs by further increasing the porosity/surface area and optimizing the pore structure is limited by the minimum pore size accessible to ions, among others, and leads to a saturation of capacitance versus surface area.26 An alternative approach to further increase the specific energy is the implementation of redox active materials. Common redox active materials used in combination with carbon are transition metal oxides, such as manganese oxide,27 ruthenium oxide,28 nickel oxide,29–34 electrochemically active polymers, like polyaniline,30–32,34,35 and polypyrrole,36–38 as well as surface functional groups, like quinones.23,39–42 Dependent on the resulting electrochemical response, we can differentiate between a battery-like or pseudocapacitive behavior.43
Manganese oxide is an attractive pseudocapacitive material because of its high redox activity, the low synthesis costs, and high availability/abundance.27,44,45 The charge storage mechanism of manganese oxide is related to faradaic surface reactions, the adsorption of positively charged electrolyte ions such as H+, Li+, Na+, and K+, but also to faradaic reactions in the bulk (cation intercalation). In these cases, redox transitions between Mn(III) and Mn(IV) states occur.27,44,46–48 The specific capacitance of manganese oxide is typically in the range of 100–200 F g−1 in neutral aqueous electrolytes. Much higher values have also been published,49 for example, up to 1380 F g−1 for thin films (5 μm)46 in the same order of magnitude than the maximum theoretical value of 1233 F g−1.50 The electrochemical performance of manganese oxide is not only determined by its morphology, phase, composition, and crystal structure,51,52 but also by thickness of the electrode/coating/particles.46,53 Although manganese oxide as supercapacitor material is already extensively studied, for example, by the groups of D. Bélanger, T. Brousse, and F. Favier,46,48,51,54–63 remaining challenges are its low structural stability, low long-time stability, small negative voltage stability, high self-discharge, and poor electrical conductivity. A more comprehensive overview about the properties of manganese oxides for supercapacitors can be found in the literature.49
The low conductivity of manganese oxide (ca. 10−5 S cm−1)64 limits the power performance. The latter can be improved by doping with metals or the hybridization with highly conductive carbon materials.49 Several carbon substrates were used in combination with different types of manganese oxide, like activated carbon fiber,24 carbon nanotubes,65–74 graphene,75–80 graphene oxide,81,82 carbon nanofoams,83,84 aerogels,83,84 nanographite sheets,85–87 carbon onions,88–90 and ordered mesoporous carbon.91–93 These carbon materials differ in crystal structure, grain/particle size, chemical composition, and oxidation state. However, the use of different electrode preparation methods, various amounts and kinds of polymer binder and conductive additive, and different electrode thicknesses make a direct comparison of electrochemical performance ratings difficult and often inconclusive. For example, Fleischmann et al. compared activated carbon and carbon onions as substrate for vanadium oxide coated with atomic layer deposition technique.94 The findings regarding the influence of high internal or external surface area on the electrochemical properties were related to the synthesis process. In a study by Gambou-Bosca et al., the difference between activated carbon and carbon black as substrates for manganese oxide was investigated.95 Yet, no comprehensive set of data exists showing the influence of the carbon substrate regarding different degrees of ordering, and carbons with high internal or external porosity. Especially when comparing organic and aqueous media, this absence complicates the development of optimization strategies for this promising class of hybrid material.
In the present study, the influence of carbon on the electrochemical performance of carbon/manganese oxide hybrid electrodes is systematically investigated for organic and aqueous media. For these two types of electrolytes, we also provide, for the first time, S-value testing to quantify the maximum width of the electrochemical voltage window. Commercially available activated carbon (AC) and nanodiamond-derived carbon onions (OLC: onion-like carbon)16 synthesized at 1300 °C and 1700 °C were hybridized with MnO2 by hydrothermal synthesis. By this way, the impact of internal vs. external surface area on the nanohybrid material performance can be investigated. Carbon onions have recently been hybridized with MnO2, but no comparison between different degrees of carbon ordering was presented.88 Our data explicitly includes activated carbon because the performance of this common type of electrode material for supercapacitors combined with manganese oxide remains ill-explored.24,62,96 Manganese oxide operated in organic and aqueous media is finally benchmarked with regard to charge and energy efficiency to assess the practicality of their implementation for energy storage applications.
2. Experimental
2.1 Materials and synthesis
Activated carbon (AC) powder YP80 was used as received (Kuraray). Carbon onions (OLC) were produced using annealing of detonation nanodiamonds (NaBond Technologies). The powder was used without further purification and placed in graphite crucibles. The typical amount of material was ∼10 g. Nanodiamond powder was annealed in a high temperature furnace (Thermal Technology) in flowing argon with a purity of 4.6 and a flow rate of 1.3 L min−1. Two annealing temperatures of 1300 °C for 10 h or 1700 °C for 1 h were used with heating and cooling rates of 20 °C min−1. The resulting carbon onions are called OLC1300 and OLC1700.Manganese oxide/carbon hybrids were produced by hydrothermal synthesis using potassium permanganate (KMnO4, Sigma Aldrich) as precursor in aqueous solution. Carbon powders (500 mg) were dispersed in 100 mL deionized water (MilliQ) via tip sonication of ∼4 W for 5 min. In a second step, potassium permanganate was added to the solution and continuously stirred and refluxed at 100 °C for 4 h until the color changed from purple to dark brown/black. Several mass ratios of KMnO4/carbon were used to reach different loadings of manganese oxide on the carbon substrate. For example, a ratio of 3:1 stands for 1.5 g of KMnO4 and 500 mg carbon powder. The nomenclature for the samples, for example “M–OLC1700 (3:1)”, includes the loading with manganese oxide (M), the type of carbon substrate (AC, OLC1300, OLC1700), and the mass ratio KMnO4/carbon (e.g., 3:1). We recently described the same synthesis route and identified the manganese oxide phase as birnessite,88 in agreement with earlier works employing permanganate as precursor.49 For simplicity, “birnessite-type manganese oxide” is shortened to “manganese oxide” in this study, although we would like to remind that the produced phase is not pure MnO2, but classifies rather as metal hydroxide with some amounts of potassium (∼12 mass%).
To investigate structure and electrochemical performance of just manganese oxide, the latter was synthesized using an adapted co-precipitation method from ref. 51. KMnO4 was dissolved in deionized water and stirred while MnCl2 was added. The mass ratio of KMnO4:MnCl2 was 2:3. The brown manganese oxide was directly collected. All samples were washed at least 3 times with deionized water, centrifuged, and dried at 90 °C and 20 mbar. Commercial MnO2 was used as received (Sigma Aldrich).
2.2 Analysis of chemical composition, structure, and porosity
The elemental composition was measured using energy-dispersive X-ray analysis (EDX) with a X-Max-150 detector from Oxford Instruments in a JSM-7500F from JEOL. The electrodes containing 10 mass% polytetrafluorethylene (PTFE) binder (see Section 2.3 for details on the electrode preparation) were placed on a carbon tape and spectra were taken at three different positions using 10 kV acceleration voltage.Transmission electron microscopy was performed with a JEOL 2100F microscope using 200 kV acceleration voltage. Powder samples were dispersed in isopropanol, tip sonicated for 10 s and drop casted on a copper grid with a lacey carbon film (Gatan).
Prior to focused ion beam (FIB) cross-sectioning in a scanning electron microscope (SEM), the samples were coated with ZnO by allowing diethyl zinc and water to react in a cyclic manner at 50 °C in a vertical-flow, hot-wall reactor (OpAL, Oxford Instruments) as described in ref. 97. This increases the contrast between pores and solid material in the SEM. Subsequently, imaging was carried out with a Zeiss Auriga 60 dual beam. The cut surface was prepared by a focused ion beam with an accelerating voltage of 30 kV and 20 pA beam current. The SEM images were acquired with the in-lens detector at 5 kV.
Thermogravimetric analysis (TGA) was used to identify the amount of manganese oxide in the hybrids. A TG 209 F1 Libra system from Netzsch was used, equipped with an automatic sample changer and alumina crucibles. Samples were heated up to 900 °C in synthetic air with a rate of 20 °C min−1 to avoid contaminations of the system. To investigate the difference in carbon ordering and phase in detail, the pure carbon powders were heated in synthetic air with a rate of 5 °C min−1. The sample mass ranged between 5 mg and 10 mg. The amount of manganese oxide was measured by the remaining sample mass after heating to 900 °C when carbon was completely oxidized.
The skeletal density was analyzed with an AccuPyc 1330 gas pycnometer (Micromeritics) using helium gas with a purity grade of 5.0. The steel crucible had a volume of 1 cm3 and the sample mass was between 100 mg and 200 mg. First, 20 purges were applied followed by 20 separate measurements. Both purge and analysis filling pressures were 134 Pa. An equilibration rate of 34 Pa min−1 was used.
Porosity and surface area were measured using nitrogen gas sorption measurements at −196 °C with an Autosorb iQ system (Quantachrome). Beforehand, the powder samples were degassed at 102 Pa and 300 °C for 24 h to remove any water residuals from the synthesis. During the measurement, the relative pressure was varied from 5 × 10−7 to 1.0 in 68 steps. The specific surface area (SSA) was calculated with the ASiQwin-software using the Brunauer Emmett Teller (BET) equation in the linear relative pressure range of 0.01–0.2.43 The DFT SSA and pore size distribution (PSD) were derived via quenched-solid DFT (QSDFT) with a slit pore shape model between 0.56 nm and 37.5 nm.46 Values for the total pore volume correspond to p/p0 = 0.95.
Raman spectroscopy was performed with a Renishaw inVia Raman Microscope equipped with a Nd-YAG laser (532 nm). For all samples a 50× objective was used with a power of 0.2 mW at the surface of the sample. The spectra were recorded with 10 accumulations and 20 s acquisition time. For each D- and G-mode we used one Voigt-profile peak and two Voigt peaks to fit the contribution of amorphous carbon (including amorphous carbon at 1520 cm−1, transpolyacetylene at ca. 1100 cm−1 and the broad signal which does not correspond to sp2-hybridized carbon). The same methodology was used in our recent study for carbon onions.98 All spectra were normalized and background subtracted with a linear baseline.
The electrode conductivity was measured with a custom built four-point probe. The diameter of the tips was 1.5 mm and the tip distance 1.75 mm. The areal resistance was measured at 5 different positions of the electrode. Using eqn (1) the electrode conductivity normalized to the thickness can be calculated.
 | (1) |
2.3 Electrochemical characterization
Free-standing and flexible PTFE-bound electrodes were used for electrochemical measurements. Carbon and hybrid powders (typically ca. 200 mg) were dispersed in ethanol and stirred in a mortar until most of the ethanol was evaporated and a carbon slurry resulted. For all samples 10 mass% PTFE (60 mass% aqueous solution, Sigma Aldrich) were added and mixed in a mortar until a dough-like carbon paste was formed. Using a rolling machine (MTI HR01, MTI Corp.) electrodes with a thickness of 60 μm (±10 μm) were rolled and dried over night at 90 °C and 20 mbar.Electrochemical characterization was carried out in 1 M LiClO4 in acetonitrile (ACN) from BASF or 1 M Na2SO4 (aqueous) from Sigma Aldrich. For all measurements, a two- and three-electrode setup was used, corresponding with full- and half-cell configuration, respectively. The electrodes were punched out to obtain discs with a diameter of 8 mm (3–5 mg) and were separated by a glass-fiber disc with diameter of 13 mm (type GF/D, GE Healthcare). The electrode/separator/electrode arrangement was compressed between two platinum disks (diameter 12 mm, thickness: 200 μm, Carl Schäfer) using spring-loaded titanium pistons, sealed by a polyether ether ketone (PEEK) body. For measurements using 1 M LiClO4 in ACN, the cells were dried at 90 °C and 20 mbar before they were put in an argon-filled glove box (MBraun Labmaster 130; O2, H2O < 1 ppm). The cells were vacuum back-filled with a syringe containing the electrolyte. The aqueous 1 M Na2SO4 electrolyte was directly vacuum filled in the cells without drying. Full-cell measurements were carried out either with a symmetric (same size, same material) or asymmetric (charge balanced counter electrode from activated carbon YP80) configuration. In a half-cell set-up, a ca. 5 times oversized AC counter electrode was used together with a reference electrode; the latter was a platinum wire for the aqueous electrolyte and PTFE-bound activated carbon YP50 from Kuraray for the organic electrolyte.
Electrochemical characterization was performed using a VSP300 and VMP300 potentiostat/galvanostat from Bio-Logic in cyclic voltammetry (CV) and galvanostatic mode with potential limitation (GCPL). The potential window for all full-cell measurements was 0.8 V in 1 M Na2SO4 and 2.2 V in 1 M LiClO4 in ACN. Half-cell measurements were used to determine the capacitance of one electrode and the stability window. For data obtained with cyclic voltammetry, a scan rate of 1–10 mV s−1 was used and the specific capacitance of two electrodes dependent on the cell voltage (cell performance) was calculated using eqn (2):
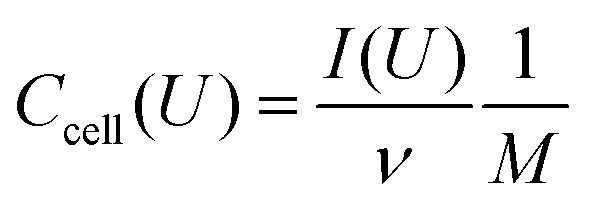 | (2) |
Galvanostatic measurements with potential limitation (GCPL) were used to calculate the capacitance for different specific currents as well as the specific energy and power. The current was normalized to the active mass and specific currents from 0.1 A g−1 to 20 A g−1 were applied. After charging the system to 0.8 V or 2.2 V, there was a 10 s resting time prior to discharge to 0 V cell voltage to enable facile assessment of the IR drop. Every GCPL cycle was repeated at least two times with 10 s of resting time in between. The specific discharge capacitance of two electrodes normalized to the active mass was calculated according to eqn (3):
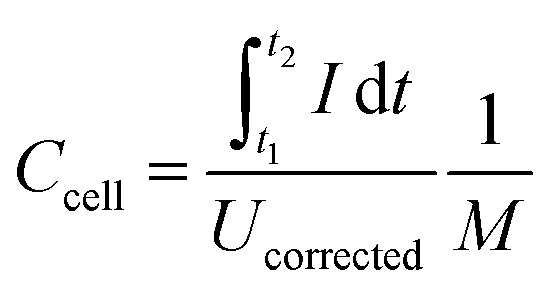 | (3) |
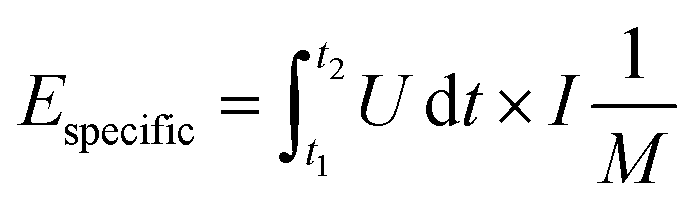 | (4) |
 | (5) |
3. Results and discussion
3.1 Morphology analysis
Two types of carbon onions (OLC) with different degrees of carbon ordering and one nanoporous activated carbon (AC) were selected for this study. TEM images of the carbon materials are shown in the ESI, Fig. S1.†Both types of carbon onions were derived from nanodiamonds by thermal annealing in argon at either 1700 °C (“high temperature”) or 1300 °C (“low temperature”). The primary particle size of carbon onions ranged between 4 nm and 10 nm featuring several carbon shells (ESI, Fig. S1A and B†). While differences in morphology were not observable in transmission electron micrographs, an increasing degree of carbon ordering for higher annealing temperatures is to be expected.98–101 In contrast to OLC, AC presented a porous structure with a high level of nanocrystalline disorder (ESI, Fig. S1C†).
Even when using the same ratio of carbon and manganese oxide, different morphologies were observed for the different carbon substrates (Fig. 1). M–OLC1700 (3:1) had a homogenous coating of the agglomerate particles (Fig. 1D and E).
 | ||
| Fig. 1 Transmission electron micrographs of the carbon/manganese oxide hybrids. (A–C) M–OLC1300 (3:1), (D–F) M–OLC1700 (3:1), (G–I) M–AC (1:2), and (J–L) M–AC (3:1). | ||
The thickness of the nanocrystalline coating was in the range of a few nanometers (∼10 nm) as determined by TEM (Fig. 1F). In contrast, M–OLC1300 (3:1) showed a fibrous coating with nanocrystalline structure (Fig. 1A–C). The skeletal density of OLC1300 (2.2 g cm−3) was 16% higher than for OLC1700 (1.9 g cm−3). Therefore, the same mass content of MnO2 would lead to a larger volumetric coating compared to the coating on the lower density substrate material. However, we see the same fibrous structure for M–OLC1300 (2:1) and (3:1) (Fig. 1A). A similar morphology was observed for M–AC (3:1) coated with the same amount of manganese oxide (Fig. 1J–L). Long and thin manganese oxide fibers grew on the surface of the activated carbon particle with a length of several hundred nanometers and a diameter of ∼20 to 50 nm (Fig. 1L). These observations align with findings in literature: depending on the reaction ratio of permanganate and carbon,96 as well as the reaction time,82 the structure of manganese oxide varies from layers and nanoparticles to nanorods with differing crystallinity. Use of a much smaller amounts of permanganate for the synthesis (M–AC (1:2)) yielded a thin coating of around 3 nm thickness, completely covering the carbon particles (Fig. 1G–I). Etching of carbon during the formation of manganese oxide is evidenced in Fig. 1C: the graphitic shells on the outside of OLC1300 were removed, while a partially amorphous and diamond-like core remained in the center of the particles. This is in accordance with results of Raman spectroscopy in Section 3.4.
The coating of carbon by manganese oxide can also be seen in cross-sectional scanning electron micrographs of the PTFE-bound electrodes (Fig. 2). The initial AC electrode shows particles in the range of several micrometers in direct contact to each other (Fig. 2A–C). Additional ZnO coating was applied to increase the contrast and allow recognition of the AC particle shape. After coating with manganese oxide (M–AC (3:1)), an additional MnO2 layer is visible partially covering the AC particles as depicted in Fig. 2D–F. Local inhomogeneous covering with MnO2 as well as the existence of a few uncoated parts of the AC particles can be the result of particle agglomeration during the wet chemical synthesis. The small size of OLC and the tendency to form agglomerates of several hundred nanometers leads to more compact electrodes (Fig. 2G). Manganese oxide deposition (M–OLC1700 (3:1)) takes place inside the agglomerates as well as on the outside, forming a homogenous and dense composite (Fig. 2J–L).
 | ||
| Fig. 2 Scanning electron micrographs of the cross sections prepared from electrodes containing 90 mass% active material (i.e., carbon and manganese oxide) and 10 mass% PTFE as binder. ZnO was deposited by means of atomic layer deposition onto the carbon particles to increase the contrast. Colored images in C, F, I, and L were used to differentiate the carbon and manganese oxide phases in B, E, H, and K. | ||
3.2 Chemical and thermogravimetric analysis
All three carbon substrates were hydrothermally hybridized with manganese oxide using KMnO4 as the precursor. This synthesis routine is well-established,49 including work by us on the high power handling ability.88 Different manganese oxide mass loadings were realized by using different carbon-to-KMnO4 ratios. The chemical composition and the manganese oxide content were measured using EDX and TGA (Table 1).| Elemental composition | Manganese oxide | ||||||
|---|---|---|---|---|---|---|---|
| C (mass%) | Mn (mass%) | O (mass%) | K (mass%) | F (mass%) | EDX (mass%) | TGA (mass%) | |
| OLC1700 | 91.8 ± 1.8 | 0 | 1.9 ± 0.5 | 0 | 6.3 ± 2.3 | 0 | 0 |
| M–OLC1700 (1:1) |
64.1 ± 1.3 | 13.9 ± 0.5 | 17.2 ± 1.9 | 4.2 ± 0.2 | 0.7 ± 0.1 | 35 ± 3 | 34 |
| M–OLC1700 (3:1) |
40.5 ± 6.6 | 27.4 ± 9.3 | 21.9 ± 6.4 | 6.8 ± 2.5 | 3.5 ± 1.8 | 56 ± 18 | 58 |
| M–OLC1700 (4:1) |
28.1 ± 1.9 | 41.0 ± 3.7 | 22.4 ± 3.4 | 7.7 ± 0.9 | 0.7 ± 0.1 | 71 ± 8 | 63 |
| M–OLC1700 (8:1) |
12.3 ± 4.3 | 47.4 ± 13.7 | 28.2 ± 11.4 | 11.0 ± 2.2 | 1.1 ± 0.4 | 87 ± 27 | 73 |
| OLC1300 | 93.6 ± 1.0 | 0 | 3.1 ± 0.1 | 0 | 3.2 ± 1.0 | 0 | 0 |
| M–OLC1300 (2:1) |
65.5 ± 8.6 | 15.5 ± 4.8 | 14.2 ± 2.5 | 3.5 ± 1.5 | 1.5 ± 0.8 | 33 ± 9 | 31 |
| M–OLC1300 (3:1) |
38.8 ± 6.9 | 22.8 ± 4.4 | 29.3 ± 3.8 | 5.3 ± 0.9 | 3.8 ± 1.3 | 57 ± 9 | 60 |
| AC | 95.5 ± 1.04 | 0 | 3.3 ± 0.5 | 0 | 1.2 ± 0.6 | 0 | 0 |
| M–AC (1:2) |
82.2 ± 32.0 | 8.9 ± 3.0 | 7.0 ± 1.3 | 1.2 ± 2.1 | 0.7 ± 0.4 | 17 ± 6 | 18 |
| M–AC (1:1) |
72.3 ± 5.8 | 12.6 ± 7.0 | 12.8 ± 3.7 | 1.9 ± 0.9 | 0.4 ± 0.2 | 27 ± 12 | 31 |
| M–AC (3:1) |
36.9 ± 7.2 | 24.9 ± 5.9 | 29.2 ± 1.3 | 6.3 ± 1.7 | 2.8 ± 2.5 | 60 ± 9 | 59 |
The chemical analysis showed fluorine in addition to Mn, O, and C, which originates from the PTFE binder. Functional groups on AC account for an oxygen content of 3.3 mass%. This value is almost identical to that of low temperature carbon onions (OLC1300:3.1 mass%); yet, the high-temperature annealing process of OLC1700 yields a lower oxygen content (1.9 mass%) in alignment with literature.98 The content of manganese oxide for the different samples is given in Table 1. By using a higher amount of permanganate for the synthesis, the manganese oxide content increased. For example, the amount of manganese oxide coating on OLC1700 varied from 35 mass% to 87 mass%. Different carbon substrates with the same amount of permanganate led to similar compositions with 56, 57, and 60 mass% manganese oxide for M–OLC1700 (3:1), M–OLC1300 (3:1), and AC (3:1).
Thermogravimetric analysis of manganese oxide showed up to ca. 120 °C an initial mass loss of around 4 mass% due to the loss of surface adsorbed water, followed by evaporation of interlayer water and dehydration of the manganese oxide until ca. 470 °C with a mass loss of ∼11 mass% (Fig. 3A).48,102 The mass loss due to progressing birnessite dehydration is dependent on the surface area and the corresponding amount of initially adsorbed water. Birnessite-type manganese oxide (KxMnO2) also undergoes a phase transformation to KxMn2O3 at around 480 °C, as seen for the carbon/manganese oxide hybrids.102 The burn-off temperatures of 631 °C for OLC1700 and 597 °C for AC were slightly higher than for OLC1300 with 566 °C, due to the higher degree of sp2-carbon ordering.
 | ||
| Fig. 3 TGA of the manganese oxide, the carbon powders, and the carbon/manganese oxide composites in synthetic air. All measurements were performed in synthetic air with a heating rate of 5 °C min−1 for the carbon powders and 20 °C min−1 for the hybrids. (A) Carbon onions and carbon onion/manganese oxide hybrids and (B) AC and AC/manganese oxide hybrids. | ||
All carbons were completely volatized between 500 °C and 700 °C. For the hybrid materials, the burn-off temperature shifts to 300–500 °C because of an enhanced etching of carbon by evaporating water, oxygen-containing gases, and catalytic effects of the manganese oxide.103,104 The remaining mass was identical to the amount of manganese oxide in the composite and was comparable to the values derived from EDX (Table 1).
3.3 Analysis of porosity, skeletal density, and electrical conductivity
Carbon onions have only outer surface area with a large fraction of mesopores (Fig. 4A), whereas activated carbon is mostly characterized by interparticle micropores (Fig. 4B). The skeletal density of both carbon onion varieties increased after adding more manganese oxide and the pore volume decreased (Fig. 4C and Table 2). The skeletal density of the AC hybrids increased from 2.2 g cm−3 to 2.4 g cm−3, while the pore volume decreased from 1.07 cm3 g−1 to 0.17 cm3 g−1 similarly to carbon onions (Fig. 4C and Table 2). This finding is in agreement with TEM micrographs, showing the homogenous covering of AC by manganese oxide and blocking the pores from the outside (Fig. 1I). Pore blocking led to a decrease of the pore volume, but not to an increase in density to the same extend because the pore walls and internal pore space are not directly coated.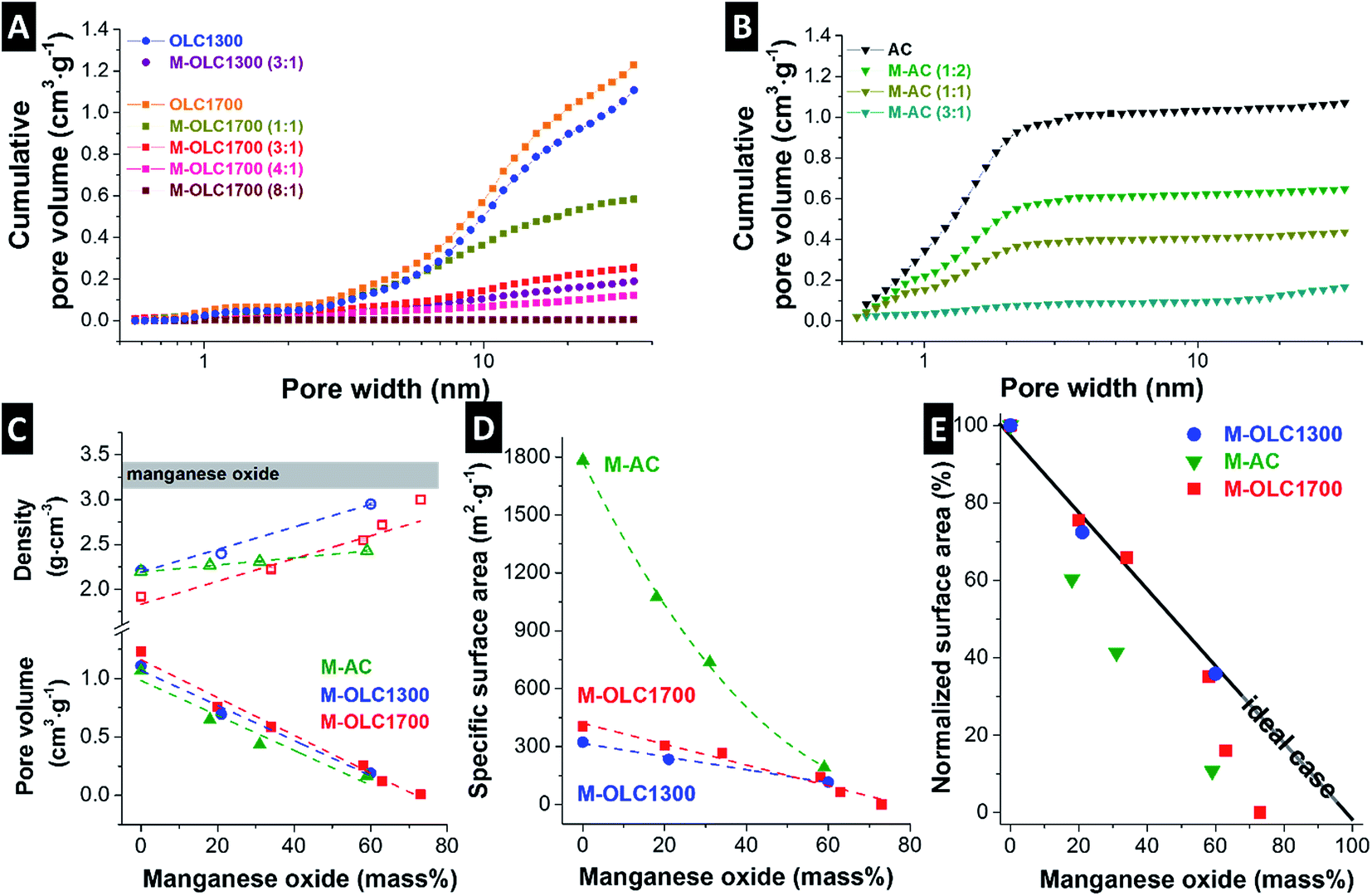 | ||
| Fig. 4 (A) Cumulative pore size distributions from nitrogen gas sorption analysis of the carbon onion samples and (B) of the AC samples. (C) Skeletal densities from helium gas pycnometry and the pore volumes from nitrogen gas sorption. (D) DFT specific surface areas from gas sorption and (E) normalized surface areas. | ||
| SSA DFT (m2 g−1) | Pore volume (cm3 g−1) | Skeletal density (g cm−3) | Conductivity (S cm−1) | |
|---|---|---|---|---|
| OLC1700 | 404 | 1.23 | 1.92 ± 0.03 | 1.78 ± 0.54 |
| M–OLC1700 (1:1) |
266 | 0.58 | 2.22 ± 0.02 | 1.98 ± 0.85 |
| M–OLC1700 (3:1) |
142 | 0.26 | 2.55 ± 0.01 | 0.09 ± 0.01 |
| M–OLC1700 (4:1) |
65 | 0.12 | 2.72 ± 0.03 | 0.02 ± 0.0 |
| M–OLC1700 (8:1) |
n.d. | 0.01 | 3.01 ± 0.04 | n.d. |
| OLC1300 | 323 | 1.11 | 2.21 ± 0.02 | 1.12 ± 0.45 |
| M–OLC1300 (2:1) |
234 | 0.70 | 2.40 ± 0.03 | n.d. |
| M–OLC1300 (3:1) |
116 | 0.19 | 2.95 ± 0.01 | n.d. |
| AC | 1783 | 1.07 | 2.20 ± 0.02 | 0.28 ± 0.03 |
| M–AC (1:2) |
1075 | 0.65 | 2.27 ± 0.03 | 0.05 ± 0.03 |
| M–AC (1:1) |
737 | 0.44 | 2.31 ± 0.01 | n.d. |
| M–AC (3:1) |
193 | 0.17 | 2.43 ± 0.01 | n.d. |
For all three carbon materials, we see a strong decrease in specific surface area (DFT) after hybridization with manganese oxide (Fig. 4D and Table 2). We determined a specific surface area of 323 m2 g−1 and 404 m2 g−1 for pristine OLC1300 and OLC1700, respectively. For a reaction ratio of (3:1), these values were reduced to 116 m2 g−1 and 142 m2 g−1, respectively. A similar value (193 m2 g−1) was measured for M–AC (3:1), which corresponds with a more than 90% decrease of the initial value (1783 m2 g−1). The composition of (3:1) was used for further comparison of the electrochemical performance because manganese oxide loading as well as surface area are comparable for the different carbon materials.
Fig. 4E shows the relative decrease of the specific surface area after manganese oxide hybridization normalized to the initial value of the carbon materials. An ideal behavior would correspond with high manganese oxide mass loading without pore blocking. This could be approached with thin coatings while maintaining the initial accessible surface area. All studied samples deviate from ideality and pore blocking occurs to a varying degree. For carbon onion hybrids, the surface structure was retained up to a maximum loading of ∼60 mass% manganese oxide due to the absence of internal pores (Fig. 2A and D),99 preventing extensive pore blocking (Fig. 4E). For higher loadings, thick coatings engulfed the agglomerates and led to a strong decrease in specific surface area. Correspondingly, interparticle pores were almost completely filled with manganese oxide, as shown in Fig. 4A. The amount of micro- and mesopores decreased for OLC samples after manganese oxide hybridization, similar to what was observed when using AC (Fig. 4B).
3.4 Raman spectroscopy
The Raman spectra of all carbon materials showed typical features of incompletely graphitized carbon, namely the D-mode at ∼1340 to 1350 cm−1, the G-mode at ∼1585 to 1600 cm−1, and a pronounced second order spectrum (2200–3500 cm−1). OLC1700 and OLC1300 showed a Raman spectrum typical for carbon onions.98,99,105–109 Both carbon onion varieties exhibited a nanocrystalline structure and a distinct contribution from amorphous carbon at ∼1520 cm−1 was seen in form of a broad transition between the D- and the G-mode. The latter was more pronounced for OLC1300 than for OLC1700 due to incomplete graphitization.98 The curved shape of the G-mode for both types of carbon onions relates to the emergence of few-layer graphene between the carbon onions as a result of the synthesis from nanodiamonds in argon.98 The Raman signal of the activated carbon presented a G-mode at 1599 cm−1 indicative of nanocrystalline carbon.110 Nanocrystallinity was also seen in TEM micrographs (ESI, Fig. S1C†), showing small crystalline domains connected by amorphous/disordered carbon forms. The amount of amorphous carbon was slightly smaller than for OLC1300 and the degree of carbon ordering in the nanoscopic domains was higher, as indicated by the smaller FWHM of the G-mode.110After hydrothermal synthesis, all samples showed additional Raman bands associated with manganese oxide between 250 cm−1 and 750 cm−1 (Fig. 5). For OLC1700, the G-mode position shifted from 1591 cm−1 to 1599 cm−1 for the highest manganese oxide loading (Fig. 5A). The redshift of the G-mode was a result of the hydrothermal synthesis, which led to superficial etched carbon particles resulting in smaller domain sizes.111 The relative increase in amorphous carbon was less than 20% for the highest loading indicative for a highly ordered material (Fig. 5E). In case of OLC1300, the change of carbon structure was not characterized by a shift of the G-mode to larger wavenumbers because the initial value was already lowered to 1599 cm−1 due to its smaller size (Fig. 5B). The relative increase in amorphous carbon neared 100% for the highest loading of manganese oxide on OLC1300 (Fig. 5E) due to the removal of graphitic outer shells and remaining diamond-like and amorphous carbon in the core (Fig. 1C). Similar to OLC1700, the etching of outer shells led to an increase of the D- and G-mode FWHM because of the combined effect of increasing defect density and decreasing domain size (Fig. 5F). The same can be seen for AC with increasing D- and G-mode FWHM, as well as increasing relative amorphous carbon content (Fig. 5C, E and F). In contrast to OLC1300, the content of amorphous carbon did not increase linearly, but reached a constant value (+60%) once a manganese oxide loading of 30% had been reached (Fig. 5E). Obviously, the comparably small external surface area of an AC particle cannot be coated with the same amount of manganese oxide than carbon onions and the impact on the carbon structure remained limited. The birnessite-type manganese oxide phase was varied as a function of the synthesis parameters. Typically, octahedral MnO6 layers present a vacancy in one over every six octahedral sites.112 Between these layers, ions (Li+, K+, Mn2+) or water molecules are intercalated depending on the synthesis and the precursor.112 Raman spectra and the frequency positions can be analyzed according to the polymerization of the MnO6 octahedra with modes at 506, 575, and 646 cm−1.112 All coated carbon onion samples showed a typical Raman spectrum of birnessite-type manganese oxide with vibrational frequencies typical for birnessite at 575 cm−1, 618 cm−1, and weak frequencies at 500 cm−1 and 730 cm−1.112 The Raman peak at 646 cm−1 is attributed to the Mn–O symmetric stretching vibration within the octahedra.
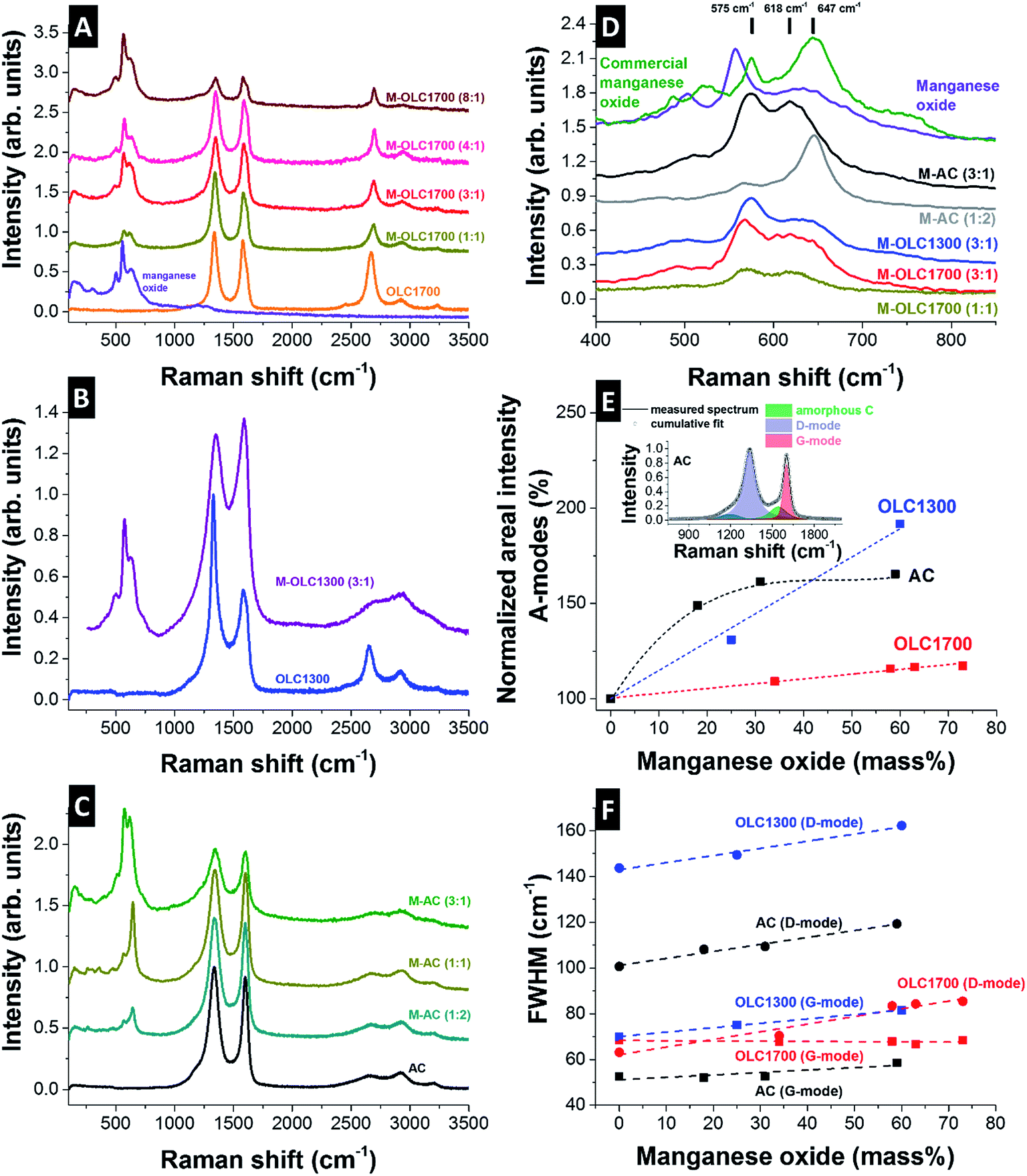 | ||
| Fig. 5 (A–C) Raman spectra of manganese oxide, the carbon powders, and the carbon/manganese oxide composites. (D) Manganese oxide Raman spectra of different hybrids. (E) Normalized areal intensity of amorphous carbon plotted against the manganese oxide content. (F) FWHM of D- and G-mode dependent on the manganese oxide loading. | ||
The lower frequency mode at 575 cm−1 comes from the stretching vibration of Mn–O in the basal plane of MnO6 sheets and is particularly strong for Mn(IV) fraction. The higher frequency mode was shifted to 618 cm−1 for carbon onions because of its strong dependence on the lattice spacing in the birnessite112 which is varied due to intercalation of potassium ions or hydration reactions.113 The insertion of ions or molecules into the lattice resulted in a local deformation, yielding shorter Mn–O bonds and partial reduction of Mn(IV) ions, leading to a blueshift in the Raman spectrum. Manganese oxide on carbon onions exhibited particularly small domain sizes of a few nanometers, as seen by TEM (Fig. 1E and F). Grain boundaries as well as distortion due to inner-grain tension may further contribute to a variation in lattice spacing and could also explain the broadness of the high frequency mode.
While no significant difference was observed for carbon onions with different manganese oxide loadings, the birnessite Raman spectrum of activated carbon hybrids showed differences dependent on the reaction ratio of permanganate to carbon. For small loadings of manganese oxide, a rather sharp high frequency mode is observed at 647 cm−1 which is close to the expected value for ideal birnessite (646 cm−1).112 Seemingly, after initial nucleation, manganese oxide layers continue to grow with rather high crystalline ordering on activated carbon. For comparison, we see much stronger nanocrystallinity with smaller manganese oxide domain sizes (ca. 2–3 nm) when using carbon onions. We conclude that carbon onions exhibit a much higher density of nucleation sites for the growth of manganese oxide. After using higher permanganate-to-activated carbon ratios, the Raman spectrum was similar to carbon onion hybrids due to the formation of nanocrystalline manganese oxide needles.
3.5 Electrochemical measurements
Electrochemistry was performed in aqueous (1 M Na2SO4) and in organic electrolyte (1 M LiClO4 in ACN). The electrochemical potential window was investigated using incremental voltage window opening in half-cells using cyclic voltammetry at 1 mV s−1 scan rate (Fig. 6A). The electrochemical stability limit (voltage window), on first approximation, can be assessed by using S-value testing, as outlined by Weingarth et al.114 S-Value testing is a quantitative measure of the irreversible charge invested during cyclic voltammetry and the limit for stable performance is conventionally set to a value of 10%. The resulting values are shown in (Fig. 6B). For the aqueous electrolyte, a potential window from −0.4 V to +0.8 V vs. platinum is seen. The cyclic voltammograms in Fig. 6A show irreversible reactions, well known for manganese oxide measured in aqueous media especially during negative polarization.60,66,115 A very high initial S-value at −0.1 V vs. Pt vanishes after conditioning (i.e., after several charge/discharge cycles). Using the organic electrolyte, a stable potential range from −1.2 V to +1.2 V vs. carbon can be identified.114 | ||
| Fig. 6 (A) Potential window opening in a half-cell using M–OLC1700 (3:1) as working electrode, 1 mV s−1 as scan rate and aqueous 1 M Na2SO4 and 1 M LiClO4 in ACN as electrolytes. (B) Corresponding S-values after Weingarth et al. (ref. 114). (C) Half-cell capacitance dependent on the manganese oxide loading. (D–G) Full-cell cyclic voltammograms at 10 mV s−1 in aqueous 1 M Na2SO4. | ||
For full-cell setups, a cell voltage of 0.8 V and 2.2 V can be used in aqueous and organic electrolyte which is also in agreement with literature.49 However, we note that it is not absolutely clear if S-value testing is the most comprehensive method to assess the maximum voltage window width for pseudocapacitive materials, because it was developed for ideal ELDC systems with the degradation mechanism related to electrolyte decomposition instead of possible electrode deterioration.114
The capacitance of an electrode material can be measured reliably using half-cells with an oversized counter electrode (activated carbon, YP80 + 5 mass% PTFE). The capacitance was measured using galvanostatic cycling to +0.5 V vs. carbon with a rate of 0.1 A g−1. By increasing the manganese oxide loading on OLC1700, the specific capacitance increased linearly until the maximum value of 150 F g−1 for 58 mass% manganese oxide (M–OLC1700 (3:1)) was reached (Fig. 6C). By further increasing the metal oxide content, the capacitance decreased. Ultimately, we see a very high electrical resistance (e.g., for M–OLC1700 (8:1)) similar to that of pure manganese oxide and no charge storage capacity was able to be determined anymore.66 The maximum value of 150 F g−1 in aqueous medium is somewhat smaller compared to other studies with maximum reported values between 200 and 400 F g−1 (Table 3). However, a fair comparison of capacitance values is only possible when similar materials and same normalizations and measurement techniques are used. Table 3 presents capacitance values of different birnessite or manganese oxide materials with KMnO4 as precursor, measured in half-cells and neutral aqueous electrolytes. Different manganese oxide contents in the composite as well as different amounts of conductive additive and binder were used. To compare the data, all capacitance values were normalized either to the MnO2 content or to the electrode without binder. After normalizing to the amount of manganese oxide, the literature values ranged from 61 to 420 F g−1. The capacitance values reported in this study are comparably high between 226 and 309 F g−1, and are attributed to the high conductivity and easy ion accessibility of the composite. The high value of 868 F g−1 reported by Makgopa et al. might stem from the use of ultrathin electrodes.88 When comparing data normalized to manganese oxide and carbon content, the capacitances are in line with most of the studies showing values in the range of 48–408 F g−1 (Table 3).
| MnO2 in the composite (mass%) | Electrolyte | Rate | Voltage window (V) | Conductive additive (mass%) | Amount of binder (mass%) | Reported capacitance (F g−1) | Normalization | Capacitance per MnO2 (F g−1) | Capacitance per electrode w/o binder (F g−1) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Birnessite on OLC | 63 | 1 M Na2SO4 | 0.1 A g−1 | 0/+0.5 | 0 | 10 | 143 | MnO2 + C | 226 | 143 | This study |
| Birnessite on OLC | 58 | 1 M LiClO4 in ACN | 0.1 A g−1 | 0/+1 | 0 | 10 | 179 | MnO2 + C | 309 | 179 | This study |
| Birnessite on OLC | 58 | 1 M Na2SO4 | 0.1 A g−1 | 0/+0.5 | 0 | 10 | 150 | MnO2 + C | 259 | 150 | This study |
| Birnessite on OLC | 34 | 1 M Na2SO4 | 0.1 A g−1 | 0/+0.5 | 0 | 10 | 83 | MnO2 + C | 244 | 83 | This study |
| Birnessite on OLC | 20 | 1 M Na2SO4 | 0.1 A g−1 | 0/+0.5 | 0 | 10 | 49 | MnO2 + C | 245 | 49 | This study |
| Birnessite on OLC | 47 | 1 M Na2SO4 | 0.1 A g−1 | 0/+0.5 | 0 | 10 | 408 | MnO2 + C | 868 | 408 | 88 |
| Birnessite nanotubes | 1 M Na2SO4 | 0.25 A g−1 | −0.2/+0.8 | 20 | 10 | 365 | MnO2 | 365 | 284 | 116 | |
| Birnessite on AC | 60 | 1 M Na2SO4 | 0.5 A g−1 | −0.2/+0.8 | 20 | 10 | 252 | MnO2 + C | 420 | 196 | 96 |
| Birnessite nanosheets | 1 M Na2SO4 | 0.3 A g−1 | −0.1/+0.9 | 20 | 10 | 269 | MnO2 | 269 | 209 | 117 | |
| Birnessite on OLC | 80 | 1 M Na2SO4 | 0.2 A g−1 | 0/+0.8 | 10 | 10 | 178 | MnO2 | 178 | 127 | 118 |
| (Na)–birnessite nanowires | 0.5 M Na2SO4 | 1 mV s−1 | 0/+1 | 10 | 10 | 191 | MnO2 | 191 | 170 | 119 | |
| Birnessite on rGO | 1 M Na2SO4 | 0.1 A g−1 | 0/+1 | 15 | 5 | 194 | Electrode | 243 | 204 | 120 | |
| Birnessite nanobelt bundle | 1 M Na2SO4 | 5 mV s−1 | −0.2/+0.8 | 15 | 5 | 268 | MnO2 | 268 | 226 | 121 | |
| Different types of birnessite | 0.1 M K2SO4 | 5 mV s−1 | 0/+0.9 | 25 | 7 | 80–110 | MnO2 | 80–110 | 58–80 | 61 | |
| Birnessite | 0.1 M Na2SO4 | 5 mV s−1 | 0/+0.9 | 15 | 5 | 150 | MnO2 | 150 | 126 | 46 | |
| Birnessite on AC fiber | 63 | 1 M Na2SO4 | 10 mA cm−2 | −0.2/+0.8 | 10 | 5 | 117 | MnO2 + C | 186 | 105 | 24 |
| Graphene-wrapped MnO2 | 91 | 1 M Na2SO4 | 0.5 A g−1 | 0/+1 | 20 | 10 | 210 | MnO2 + C | 231 | 180 | 122 |
| Nanostructured MnO2 | 1 M Na2SO4 | 5 mV s−1 | −0.2/+0.8 | 20 | 5 | 72–168 | MnO2 | 72–168 | 57–133 | 71 | |
| Nanostructured MnO2 | 0.1 M Na2SO4 | 0.5 mA cm−2 | 0/+1 | 20 | 5 | 61–250 | MnO2 | 61–250 | 48–133 | 123 | |
| Nanostructured α-MnO2 | 0.1 M Na2SO4 | 0.5 mA cm−2 | 0/+1 | 20 | 10 | 297 | MnO2 | 297 | 231 | 124 | |
| Amorphous MnO2 + CB | 60–80 | 0.65 M K2SO4 | 2 mV s−1 | −0.1/+0.9 | 0 | 10 | 100–141 | Electrode | 164–175 | 111–157 | 95 |
| Amorphous MnO2 + AC | 60–80 | 0.65 M K2SO4 | 2 mV s−1 | −0.1/+0.9 | 0 | 10 | 134–150 | Electrode | 174–183 | 149–167 | 95 |
| Amorphous MnO2 + AC | 60 | 0.65 M K2SO4 | 2 mV s−1 | 0/+0.9 | 0 | 10 | 129 | Electrode | 239 | 143 | 62 |
| Amorphous MnO2 on AC | 60 | 0.65 M K2SO4 | 2 mV s−1 | 0/+0.9 | 0 | 10 | 122 | Electrode | 227 | 136 | 62 |
Fig. 6D depicts CVs of the full-cells of OLC1700. The rectangular shape of OLC1700 demonstrates nearly ideal double-layer capacitor behavior, in alignment with literature.16,17 By increasing the metal oxide loading, the cell capacitance increased by maintaining the quasi rectangular shape, which evidences the highly pseudocapacitive behavior of the hybrid electrodes.43,44,46 With a loading of 73 mass% manganese oxide, the resistivity of the electrode was too high and no significant capacitance could be measured (Fig. 6D and Table 2). The conductivity of the electrodes followed the same trend with 1.78 ± 0.54 S cm−1 for OLC1700, 1.98 ± 0.85 S cm−1 for M–OLC1700 (1:1), and decreasing values until the conductivity for the highest loading was smaller than the detection limit (Table 2). By adding 10 mass% OLC1700 as conductive additive during electrode fabrication to the material (M–OLC1700 (8:1) + 10% OLC), the conductivity of the electrode was enhanced to 1.52 ± 0.44 S cm−1, leading to a pronouncedly rectangular shape of the CV (Fig. 6E).
During the synthesis, manganese oxide first nucleates on the carbon substrate leading to a thin layer with low resistivity and rather low electrode capacitance (M–OLC1700 (1:1)). Further coating increases the layer thickness until a balance of electrical conductivity and electrochemical response (i.e., maximum capacitance) is reached (M–OLC1700 (3:1)). As this process continues, the conductivity decreases significantly (M–OLC1700 (8:1)) (Table 2). This is also supported by the growth of manganese oxide needles which are not directly in contact with conductive carbon. Adding carbon onions as conductive additive between the non-conductive parts of thick layer or needle-like manganese oxide (M–OLC1700 (8:1) + 10% OLC) is a facile approach to significantly increase conductivity and capacitance (Table 2). The direct influence of conductivity and electron support of the manganese oxide by the conductive carbon is supported by the low performance of OLC1300 in the composite (Fig. 6D).
Low temperature carbon onions (OLC1300) present a lower degree of carbon ordering than OLC1700 (Fig. 5D), explaining the lower electrode conductivity of 1.12 ± 0.45 S cm−1 and inferior rate handling.98,100,101,125 The cyclic voltammogram of OLC1300 is similar to OLC1700 with a nearly perfect rectangular shape (Fig. 6D). After adding manganese oxide, the capacitive behavior vanished. Even for very small manganese oxide loadings, the high resistivity (Table 2) of the electrode prevents the emergence of a characteristic pseudocapacitive behavior and results in negligible capacitance. As shown by Raman spectroscopy and TEM analysis, the hydrothermal synthesis of manganese oxide coating on carbon involves etching of outer graphitic layers, resulting in less conductive amorphous carbon cores. Consequently, the electrochemical performance of carbon/manganese oxide hybrids is directly influenced by the degree of carbon ordering and the electrical conductivity, since all other parameters were kept constant.
For AC, using the same synthesis, no significant increase in capacitance was observed after variation of the manganese oxide loading (Fig. 6B and E). The highest value of 96 F g−1 at 0.5 V in a half-cell is seen for the initial AC without additional redox activity and 84 F g−1 for M–AC (1:2). In recent literature, somewhat larger values of 117 F g−1 (ref. 24) and 252 F g−1 (ref. 96) were reported for activated carbon (fiber)/manganese oxide composites. Coating of the porous particles with a dense layer of metal oxide resulted in pore blocking, leading to a decrease in surface area as shown by TEM, gas sorption, and gas pycnometry. Still, for thin layers (i.e., M–AC (1:2) and M–AC (1:1)), the capacitance was similar to the initial AC, even if the surface area decreases to ∼60% of the initial value (731 m2 g−1). Cyclic voltammograms indicate a sufficiently high conductivity (Table 2) in comparison with OLC1300/manganese oxide hybrids and a rectangular shape (Fig. 6E). For higher manganese oxide loadings, the capacitance quickly drops and the electrochemical response becomes more resistive. The layer thickness as well as the growth of needles starts at much lower amounts of manganese oxide precursor compared to OLC. For M–OLC1700 (3:1), the best performance was measured with the highest capacitance of 150 F g−1, whereas the same amount of metal oxide with AC as substrate showed a negligible capacitance (ca. 10 F g−1).
Coating thickness,46,53 morphology,71 and structure51 of the manganese oxide phase not only influence the (equilibrium = low-rate) capacitance, but also result in differing power performances. Fig. 7A and B present the rate handling by applying different current densities in full-cell mode. In agreement with literature, the rate behavior of OLC was demonstrated to be superior to AC with a capacitance retention of more than 80% at 20 A g−1 compared to less than 40% at 10 A g−1.101 For M–AC (1:2) with a surface area > 1000 m2 g−1, a significant contribution of double-layer capacitance can be assumed. However, the thin and highly conductive coating enabled a very fast system response due to fast surface redox and intercalation effects. Therefore, OLC1700 hybrids resulted in a higher capacitance and in a better rate handling due to higher conductivity and highly accessible outer surface area (Fig. 7A and B). For M–OLC1700 (8:1), thick metal oxide layers resulted in high contact resistances between the particles and less facile electron support. After adding 10 mass% OLC (M–OLC1700 (8:1) + 10 mass% OLC), the conductivity of the manganese oxide surface coating was increased. M–OLC1700 (3:1) with a manganese oxide content of 58 mass% exhibited the best performance with the highest measured capacitance of 150 F g−1 and good rate handling. However, the specific energy for 0.8 V cell voltage in aqueous electrolyte results in only 2.8 W h kg−1, 40% larger than for AC (∼2 W h kg−1). To further improve the specific energy, the maximum cell voltage can be increased when using an organic electrolyte instead of an aqueous medium. In our case, we investigated 1 M LiClO4 in ACN. The capacitance of M–OLC1700 (3:1) was increased to 176 F g−1 at 0.5 V and 179 F g−1 at 1 V and 0.1 A g−1 (measured in a half-cell). Corresponding cyclic voltammograms for the full-cell setup are given in Fig. 7C. The extended voltage window translates to an enhanced specific energy of 16.4 W h kg−1 which is more than 5-times higher than in aqueous media.
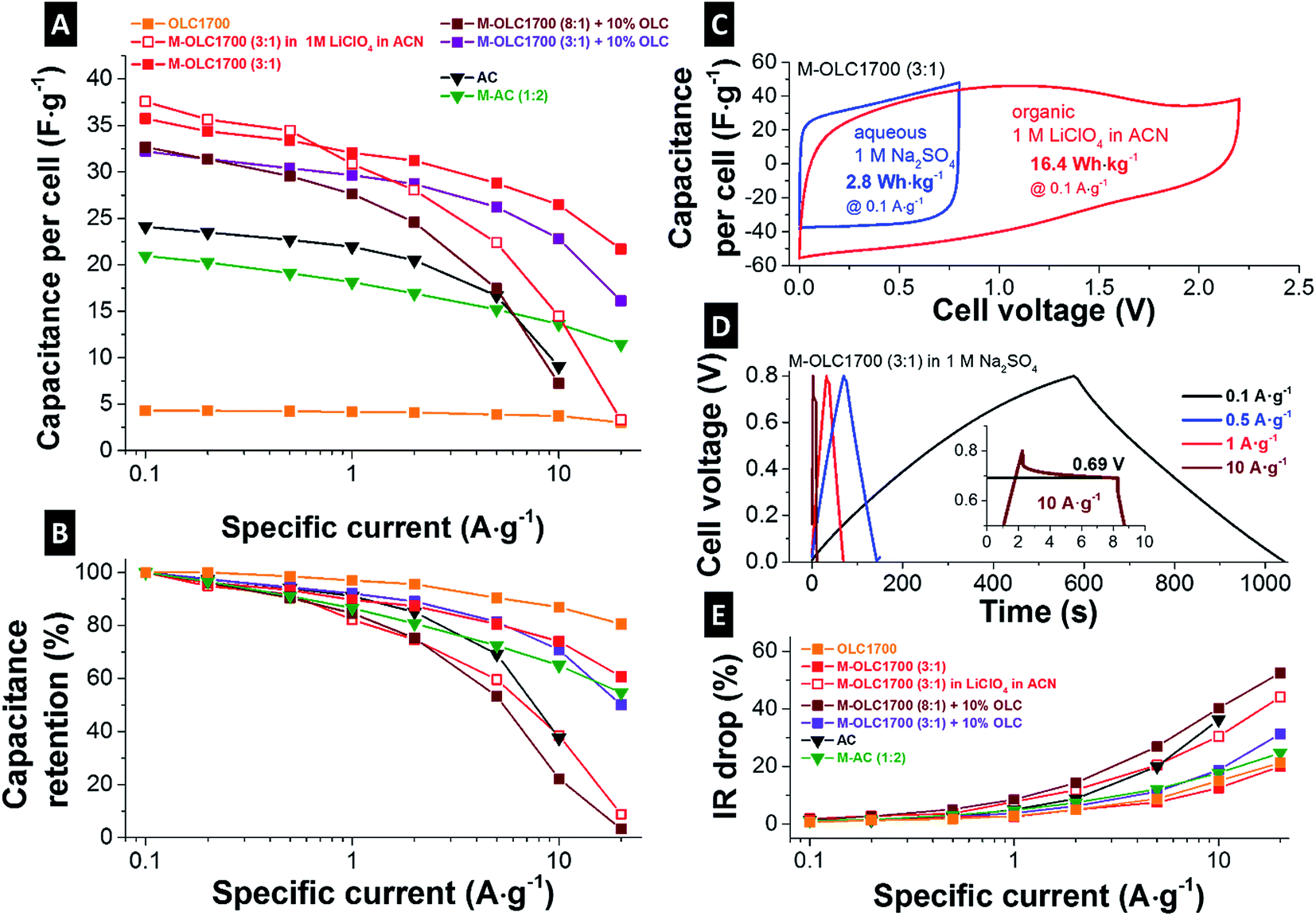 | ||
| Fig. 7 (A) Rate handling plot: full-cell capacitance dependent on the applied current density. (B) Normalized rate handling plot. (C) Cyclic voltammograms of M–OLC1700 (3:1) in full-cell mode using aqueous 1 M Na2SO4 and 1 M LiClO4 in ACN. (D) Galvanostatic cycling plots of M–OLC1700 (3:1) aqueous 1 M Na2SO4 using different specific currents. The inset shows the IR drop by applying a specific current of 10 A g−1. (E) Normalized IR drop: normalized to IR drop at 0.1 A g−1 (0.8 V and 2.2 V for aqueous and organic electrolyte). All measurements were done in aqueous 1 M Na2SO4 electrolyte if not otherwise declared. | ||
Up to a specific current of 1 A g−1, the cell capacitance in the organic medium was comparable to the same material operated in the aqueous electrolyte (Fig. 7A and B). For higher rates, the capacitance retention dropped more severely compared to aqueous medium. The normalized IR drop is shown in Fig. 7E and the corresponding charge–discharge curves in Fig. 7D present higher values in organic medium compared to the aqueous electrolyte. For electrodes with higher conductivity (Table 2), the IR drop was smaller and the resulting cell voltage larger (Fig. 7E). The initial carbon onions (OLC1700) presented an increase in IR drop of ∼20% when increasing the specific current from 0.1 A g−1 to 20 A g−1 due to high conductivity and high ion accessibility. The same was measured for M–OLC1700 (3:1) with a thin manganese oxide coating.
Electrochemical energy storage devices can be compared conveniently via Ragone plots.126 Fig. 8A shows the specific energy of the cell normalized to the active mass of both electrodes versus the specific power derived from galvanostatic cycling by using different rates. Carbon onions (OLC1700) present excellent specific power of more than 10 kW kg−1, but with a comparable low specific energy of less than 0.4 W h kg−1 in aqueous electrolyte. Values for the specific energy were at 2.8 W h kg−1 for M–OLC1700 (3:1), which is larger than activated carbon with ∼2 W h kg−1. The energy capacity can be increased when using organic electrolyte with 2.2 V cell voltage to 16.4 W h kg−1. By adding carbon onions as conductive additive, the specific power can be slightly increased for the manganese oxide hybrid (M–OLC1700 (3:1) + 10 mass% OLC), but the specific energy is decreased due to smaller capacitance. Additional manganese oxide loading (M–OLC1700 (8:1) + 10 mass% OLC) decreased the specific power as well as the specific energy.
 | ||
| Fig. 8 (A) Ragone plot derived from galvanostatic cycling with potential limitation using 0.8 V and 2.2 V cell voltage for aqueous and organic electrolyte. All values are normalized to two electrodes. (B) Charge efficiency and (C) energy efficiency dependent on the applied specific currents. (D) Long-time stability test using 10000 galvanostatic cycles using symmetric and asymmetric full-cells. All measurements were done in aqueous 1 M Na2SO4 electrolyte if not otherwise stated. | ||
The optimum performance was found for 58 mass% manganese oxide, which is in agreement with recent studies using similar amounts.24,88,96 Good performance at low and high rates is important, but only when also sufficient long-time stability is maintained. For manganese oxide in organic and neutral aqueous media, the potential window in the negative range is limited to −1.2 V vs. carbon and −0.4 V vs. Pt (Fig. 6A and B). For aqueous media, the stability is rather low due to the dissolution of manganese oxide in the negative range.60,66,115 This potential-dependent stability makes manganese oxide an attractive electrode material for positive polarization, but not for the negative side in aqueous media.44 Although a small cell voltage of 0.8 V for aqueous electrolyte and 2.2 V for organic electrolyte was used, the electrodes lost more than 40% of capacitance after 10000 galvanostatic cycles, in agreement with the measured stability range in Fig. 6A and B. Even in organic electrolyte at 2.2 V, the material had a capacitance retention of less than 40%. This clearly shows that S-value stability testing for pseudocapacitive materials, including faradaic reactions, may overestimate the actual potential windows.
For low rates and high loadings, the charge efficiency falls below 80% (Fig. 8B). Irreversible reactions take place, for example influenced by electrode dissolution like the Mn(IV) reduction, whereas the charge efficiency for the pristine carbon powders remains constant. For higher rates, these reactions still occur, but are not visible in the electrochemical data because of the high currents related to fast reactions. Taking into account the increase in IR drop in Fig. 7E and the charge efficiency (as a result of irreversible reactions; e.g., dissolution of the material) in Fig. 8B, the change of energy efficiency (Fig. 8C) can be explained. The latter shows low values for low and high rates coming from a voltage loss at high rates and low charge efficiency at low rates.
The low stability of manganese oxide in the negative range can be avoided by using an asymmetric fabrication technique.44,59,60,115 In this study, a charge balanced AC counter electrode was used (negative polarization) and the carbon onion/manganese oxide hybrid (positive polarization). Fig. 8D demonstrates the improved long-time stability of asymmetric devices in aqueous and organic electrolyte, with a capacitive retention of around 72% after 10000 charge/discharge cycles. Clearly, the instability of manganese oxide remains a key challenge for further adaption of the technology, but asymmetric device architectures can greatly improve the performance stability.
4. Conclusions
This study provides a comprehensive data set of hydrothermally synthesized birnessite-type manganese oxide on different carbon substrates for supercapacitor applications. As observable in our data, the metal oxide coatings block the internal pores of activated carbon. Depending on the amount of manganese oxide loading, its specific surface area decreased by more than 80%. The very low external surface area of AC did not provide enough active sites for the reaction with permanganate (i.e., the manganese oxide precursor), so that needle-like structures with high resistivity form which protrude into the interparticle space of the electrode. For comparison, high temperature carbon onions, with accessible surface area only on the outer side were homogenously coated, resulting in a strong increase in capacitance and overall good rate behavior with ∼75% capacitance retention at 10 A g−1 applied specific current.Besides porosity and morphology of the carbon substrate, also the degree of carbon ordering strongly influences the electrochemical performance of carbon/manganese oxide hybrids. High and low temperature carbon onions (OLC1700 and OLC1300) with different degrees of carbon ordering were compared. The lower conductivity of OLC1300 was found to be insufficient to overcome the low conductivity of manganese oxide, resulting in a very high resistance of the system. For OLC1700, a strong increase in capacitance from ∼20 F g−1 to 150 F g−1 was measured with a specific energy of 2.8 W h kg−1 in aqueous and 16.4 W h kg−1 in organic electrolyte, while showing a good rate handling behavior. The limited long-time stability, a well-known problem of manganese oxide, was strongly improved by building asymmetric full-cells with activated carbon on the negative side. However, the stability remains a key challenge for further advancements of the technology.
Acknowledgements
This work was supported by the CREATe-Network Project, Horizon 2020 of the European Commission (RISE Project No. 644013). This work was carried out within the framework of the graduate course “Materials for the efficient use of energy” at Saarland University. The authors thank Prof. Eduard Arzt (INM) for his continuing support. Nicolas Jäckel (INM) is thanked for initiating collaboration with the Thiele Group and for helpful comment and discussions. We thank Jan Laube and Prof. Zacharias from the Laboratory for Nanotechnology (University of Freiburg) for the atomic layer deposition of ZnO.References
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- J. B. Goodenough, Energy Environ. Sci., 2014, 7, 14–18 CAS.
- Z. Yang, J. Zhang, M. C. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- N. S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y. K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science Magazine, 2014, p. 343 Search PubMed.
- E. Frackowiak and F. Beguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- N. Jäckel, M. Rodner, A. Schreiber, J. Jeongwook, M. Zeiger, M. Aslan, D. Weingarth and V. Presser, J. Power Sources, 2016, 326, 660–671 CrossRef.
- D. Qu and H. Shi, J. Power Sources, 1998, 74, 99–107 CrossRef CAS.
- H. Shi, Electrochim. Acta, 1996, 41, 1633–1639 CrossRef CAS.
- Y. Gogotsi, A. Nikitin, H. Ye, W. Zhou, J. E. Fischer, B. Yi, H. C. Foley and M. W. Barsoum, Nat. Mater., 2003, 2, 591–594 CrossRef CAS PubMed.
- A. Jänes, L. Permann, M. Arulepp and E. Lust, Electrochem. Commun., 2004, 6, 313–318 CrossRef.
- V. Presser, L. Zhang, J. J. Niu, J. McDonough, C. Perez, H. Fong and Y. Gogotsi, Adv. Energy Mater., 2011, 1, 423–430 CrossRef CAS.
- A. Fuertes, G. Lota, T. Centeno and E. Frackowiak, Electrochim. Acta, 2005, 50, 2799–2805 CrossRef CAS.
- M. Zeiger, N. Jäckel, V. N. Mochalin and V. Presser, J. Mater. Chem. A, 2016, 4, 3172–3196 CAS.
- M. E. Plonska-Brzezinska and L. Echegoyen, J. Mater. Chem. A, 2013, 1, 13703–13714 CAS.
- A. Bello, K. Makgopa, M. Fabiane, D. Dodoo-Ahrin, K. I. Ozoemena and N. Manyala, J. Mater. Sci., 2013, 48, 6707–6712 CrossRef CAS.
- C. J. Jafta, F. Nkosi, L. le Roux, M. K. Mathe, M. Kebede, K. Makgopa, Y. Song, D. Tong, M. Oyama and N. Manyala, Electrochim. Acta, 2013, 110, 228–233 CrossRef CAS.
- E. Frackowiak, K. Jurewicz, S. Delpeux and F. Beguin, J. Power Sources, 2001, 97, 822–825 CrossRef.
- E. Frackowiak, K. Metenier, V. Bertagna and F. Beguin, Appl. Phys. Lett., 2000, 77, 2421–2423 CrossRef CAS.
- E. Ra, E. Raymundo-Piñero, Y. Lee and F. Béguin, Carbon, 2009, 47, 2984–2992 CrossRef CAS.
- M. Zeiger, D. Weingarth and V. Presser, ChemElectroChem, 2015, 2, 1117–1127 CrossRef CAS.
- K. Zhu, Y. Wang, J. A. Tang, H. Qiu, X. Meng, Z. Gao, G. Chen, Y. Wei and Y. Gao, RSC Adv., 2016, 6, 14819–14825 RSC.
- U. Fischer, R. Saliger, V. Bock, R. Petricevic and J. Fricke, J. Porous Mater., 1997, 4, 281–285 CrossRef CAS.
- O. Barbieri, M. Hahn, A. Herzog and R. Kötz, Carbon, 2005, 43, 1303–1310 CrossRef CAS.
- C. Xu, F. Kang, B. Li and H. Du, J. Mater. Res., 2010, 25, 1421–1432 CrossRef CAS.
- J. Zheng, P. Cygan and T. Jow, J. Electrochem. Soc., 1995, 142, 2699–2703 CrossRef CAS.
- M. E. Plonska-Brzezinska, D. M. Brus, A. Molina-Ontoria and L. Echegoyen, RSC Adv., 2013, 3, 25891–25901 RSC.
- W.-C. Chen and T.-C. Wen, J. Power Sources, 2003, 117, 273–282 CrossRef CAS.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L.-C. Qin, J. Phys. Chem. C, 2011, 115, 23584–23590 CAS.
- I. Kovalenko, D. G. Bucknall and G. Yushin, Adv. Funct. Mater., 2010, 20, 3979–3986 CrossRef CAS.
- M. E. Plonska-Brzezinska, J. Mazurczyk, B. Palys, J. Breczko, A. Lapinski, A. T. Dubis and L. Echegoyen, Chem.–Eur. J., 2012, 18, 2600–2608 CrossRef CAS PubMed.
- H. Zengin, W. Zhou, J. Jin, R. Czerw, D. W. Smith, L. Echegoyen, D. L. Carroll, S. H. Foulger and J. Ballato, Adv. Mater., 2002, 14, 1480–1483 CrossRef CAS.
- K.-S. Kim and S.-J. Park, J. Solid State Electrochem., 2012, 16, 2751–2758 CrossRef CAS.
- L.-Z. Fan and J. Maier, Electrochem. Commun., 2006, 8, 937–940 CrossRef CAS.
- K. Jurewicz, S. Delpeux, V. Bertagna, F. Béguin and E. Frackowiak, Chem. Phys. Lett., 2001, 347, 36–40 CrossRef CAS.
- O. Mykhailiv, M. Imierska, M. Petelczyc, L. Echegoyen and M. E. Plonska-Brzezinska, Chem.–Eur. J., 2015, 21, 5783–5793 CrossRef CAS PubMed.
- D. M. Anjos, J. K. McDonough, E. Perre, G. M. Brown, S. H. Overbury, Y. Gogotsi and V. Presser, Nano Energy, 2013, 2, 702–712 CrossRef CAS.
- S. Bailey, I. Ritchie, N. Searcy and P. Singh, J. Appl. Electrochem., 1988, 18, 368–373 CrossRef CAS.
- X. Chen, H. Wang, H. Yi, X. Wang, X. Yan and Z. Guo, J. Phys. Chem. C, 2014, 118, 8262–8270 CAS.
- A. Le Comte, D. Chhin, A. Gagnon, R. Retoux, T. Brousse and D. Belanger, J. Mater. Chem. A, 2015, 3, 6146–6156 CAS.
- T. Brousse, D. Bélanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef CAS.
- D. Bélanger, L. Brousse and J. W. Long, Electrochem. Soc. Interface, 2008, 17, 49 Search PubMed.
- L. Pan, K.-X. Wang, X.-D. Zhu, X.-M. Xie and Y.-T. Liu, J. Mater. Chem. A, 2015, 3, 6477–6483 CAS.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2004, 16, 3184–3190 CrossRef CAS.
- T.-H. Wu, D. Hesp, V. R. Dhanak, C. Collins, F. Braga, L. Hardwick and C.-C. Hu, J. Mater. Chem. A, 2015, 3, 12786–12795 CAS.
- L. Athouël, F. Moser, R. Dugas, O. Crosnier, D. Bélanger and T. Brousse, J. Phys. Chem. C, 2008, 112, 7270–7277 Search PubMed.
- W. Wei, X. Cui, W. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40, 1697–1721 RSC.
- A. Gambou-Bosca and D. Bélanger, ECS Trans., 2015, 64, 13–20 CrossRef CAS.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2002, 14, 3946–3952 CrossRef CAS.
- O. Ghodbane, J.-L. Pascal, B. Fraisse and F. Favier, ACS Appl. Mater. Interfaces, 2010, 2, 3493–3505 CAS.
- M. Wu, L. Zhang, J. Gao, C. Xiao, D. Wang, A. Chen and S. Zhang, J. Electroanal. Chem., 2008, 613, 125–130 CrossRef CAS.
- T. Brousse, P.-L. Taberna, O. Crosnier, R. Dugas, P. Guillemet, Y. Scudeller, Y. Zhou, F. Favier, D. Bélanger and P. Simon, J. Power Sources, 2007, 173, 633–641 CrossRef CAS.
- O. Ghodbane, F. Ataherian, N.-L. Wu and F. Favier, J. Power Sources, 2012, 206, 454–462 CrossRef CAS.
- O. Ghodbane, J.-L. Pascal and F. Favier, ACS Appl. Mater. Interfaces, 2009, 1, 1130–1139 CAS.
- D. J. Jones, E. Wortham, J. Roziere, F. Favier, J.-L. Pascal and L. Monconduit, J. Phys. Chem. Solids, 2004, 65, 235–239 CrossRef CAS.
- Y. Lei, C. Fournier, J.-L. Pascal and F. Favier, Microporous Mesoporous Mater., 2008, 110, 167–176 CrossRef CAS.
- L. Athouël, F. Moser, R. Dugas, O. Crosnier, D. Bélanger and T. Brousse, ECS Trans., 2008, 16, 119–123 Search PubMed.
- T. Brousse, M. Toupin and D. Belanger, J. Electrochem. Soc., 2004, 151, A614–A622 CrossRef CAS.
- T. Brousse, M. Toupin, R. Dugas, L. Athouël, O. Crosnier and D. Bélanger, J. Electrochem. Soc., 2006, 153, A2171–A2180 CrossRef CAS.
- A. Gambou-Bosca and D. Bélanger, J. Electrochem. Soc., 2015, 162, A5115–A5123 CrossRef CAS.
- C. Ramirez-Castro, O. Crosnier, L. Athouël, R. Retoux, D. Bélanger and T. Brousse, J. Electrochem. Soc., 2015, 162, A5179–A5184 CrossRef CAS.
- T. M. McEvoy, J. W. Long, T. J. Smith and K. J. Stevenson, Langmuir, 2006, 22, 4462–4466 CrossRef CAS PubMed.
- V. Subramanian, H. Zhu and B. Wei, Electrochem. Commun., 2006, 8, 827–832 CrossRef CAS.
- E. Raymundo-Pinero, V. Khomenko, E. Frackowiak and F. Beguin, J. Electrochem. Soc., 2005, 152, A229–A235 CrossRef CAS.
- C. Y. Lee, H. M. Tsai, H. J. Chuang, S. Y. Li, P. Lin and T. Y. Tseng, J. Electrochem. Soc., 2005, 152, A716–A720 CrossRef CAS.
- Y.-T. Wu and C.-C. Hu, J. Electrochem. Soc., 2004, 151, A2060–A2066 CrossRef CAS.
- S.-B. Ma, K.-Y. Ahn, E.-S. Lee, K.-H. Oh and K.-B. Kim, Carbon, 2007, 45, 375–382 CrossRef CAS.
- J. Li and I. Zhitomirsky, J. Mater. Process. Technol., 2009, 209, 3452–3459 CrossRef CAS.
- V. Subramanian, H. Zhu, R. Vajtai, P. Ajayan and B. Wei, J. Phys. Chem. B, 2005, 109, 20207–20214 CrossRef CAS PubMed.
- Z. Fan, J. Chen, B. Zhang, F. Sun, B. Liu and Y. Kuang, Mater. Res. Bull., 2008, 43, 2085–2091 CrossRef CAS.
- S.-L. Chou, J.-Z. Wang, S.-Y. Chew, H.-K. Liu and S.-X. Dou, Electrochem. Commun., 2008, 10, 1724–1727 CrossRef CAS.
- Z. Fan, J. Chen, B. Zhang, B. Liu, X. Zhong and Y. Kuang, Diamond Relat. Mater., 2008, 17, 1943–1948 CrossRef CAS.
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020–4028 CrossRef CAS PubMed.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 3825–3833 CrossRef CAS.
- Z. Li, Y. Mi, X. Liu, S. Liu, S. Yang and J. Wang, J. Mater. Chem., 2011, 21, 14706–14711 RSC.
- Z. Fan, J. Yan, T. Wei, L. Zhi, G. Ning, T. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366–2375 CrossRef CAS.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L.-C. Qin, Carbon, 2011, 49, 2917–2925 CrossRef CAS.
- Y. He, W. Chen, X. Li, Z. Zhang, J. Fu, C. Zhao and E. Xie, ACS Nano, 2012, 7, 174–182 CrossRef PubMed.
- A. Sumboja, C. Y. Foo, X. Wang and P. S. Lee, Adv. Mater., 2013, 25, 2809–2815 CrossRef CAS PubMed.
- S. Chen, J. Zhu, X. Wu, Q. Han and X. Wang, ACS Nano, 2010, 4, 2822–2830 CrossRef CAS PubMed.
- A. E. Fischer, K. A. Pettigrew, D. R. Rolison, R. M. Stroud and J. W. Long, Nano Lett., 2007, 7, 281–286 CrossRef CAS PubMed.
- J. W. Long, M. B. Sassin, A. E. Fischer, D. R. Rolison, A. N. Mansour, V. S. Johnson, P. E. Stallworth and S. G. Greenbaum, J. Phys. Chem. C, 2009, 113, 17595–17598 CAS.
- C. Wan, K. Azumi and H. Konno, J. Appl. Electrochem., 2007, 37, 1055–1061 CrossRef CAS.
- C. Wan, K. Azumi and H. Konno, Electrochim. Acta, 2007, 52, 3061–3066 CrossRef CAS.
- J. Yan, Z. Fan, T. Wei, Z. Qie, S. Wang and M. Zhang, Mater. Sci. Eng., B, 2008, 151, 174–178 CrossRef CAS.
- K. Makgopa, P. M. Ejikeme, C. J. Jafta, K. Raju, M. Zeiger, V. Presser and K. I. Ozoemena, J. Mater. Chem. A, 2015, 3, 3480–3490 CAS.
- R. Borgohain, J. Li, J. P. Selegue and Y.-T. Cheng, J. Phys. Chem. C, 2012, 116, 15068–15075 CAS.
- R. Borgohain, J. P. Selegue and Y.-T. Cheng, J. Mater. Chem. A, 2014, 2, 20367–20373 CAS.
- X. Dong, W. Shen, J. Gu, L. Xiong, Y. Zhu, H. Li and J. Shi, J. Phys. Chem. B, 2006, 110, 6015–6019 CrossRef CAS PubMed.
- L. L. Zhang, T. Wei, W. Wang and X. Zhao, Microporous Mesoporous Mater., 2009, 123, 260–267 CrossRef CAS.
- S. Yoon, C. Lee, S. M. Oh, Y.-K. Park and W. C. Choi, J. Non-Cryst. Solids, 2009, 355, 252–256 CrossRef CAS.
- S. Fleischmann, N. Jäckel, M. Zeiger, B. Krüner, I. Grobelsek, P. Formanek, S. Choudhury, D. Weingarth and V. Presser, Chem. Mater., 2016, 28, 2802–2813 CrossRef CAS.
- A. Gambou-Bosca and D. Bélanger, J. Mater. Chem. A, 2014, 2, 6463–6473 CAS.
- X. Zhang, X. Sun, H. Zhang, D. Zhang and Y. Ma, Mater. Chem. Phys., 2012, 137, 290–296 CrossRef CAS.
- S. Vierrath, F. Güder, A. Menzel, M. Hagner, R. Zengerle, M. Zacharias and S. Thiele, J. Power Sources, 2015, 285, 413–417 CrossRef CAS.
- M. Zeiger, N. Jäckel, D. Weingarth and V. Presser, Carbon, 2015, 94, 507–517 CrossRef CAS.
- M. Zeiger, N. Jäckel, M. Aslan, D. Weingarth and V. Presser, Carbon, 2015, 84, 584–598 CrossRef CAS.
- D. Weingarth, M. Zeiger, N. Jäckel, M. Aslan, G. Feng and V. Presser, Adv. Energy Mater., 2014, 4, 1400316 CrossRef.
- J. K. McDonough, A. I. Frolov, V. Presser, J. Niu, C. H. Miller, T. Ubieto, M. V. Fedorov and Y. Gogotsi, Carbon, 2012, 50, 3298–3309 CrossRef CAS.
- S. Bach, J. Pereira-Ramos, N. Baffier and R. Messina, Electrochim. Acta, 1991, 36, 1595–1603 CrossRef CAS.
- B. Shen, Energy Convers. Manage., 2006, 47, 1429–1437 CrossRef CAS.
- Y. Peng, Z. Chen, J. Wen, Q. Xiao, D. Weng, S. He, H. Geng and Y. Lu, Nano Res., 2011, 4, 216–225 CrossRef CAS.
- O. O. Mykhaylyk, Y. M. Solonin, D. N. Batchelder and R. Brydson, J. Appl. Phys., 2005, 97, 074302 CrossRef.
- E. D. Obraztsova, M. Fujii, S. Hayashi, V. L. Kuznetsov, Y. V. Butenko and A. L. Chuvilin, Carbon, 1998, 36, 821–826 CrossRef CAS.
- D. Roy, M. Chhowalla, H. Wang, N. Sano, I. Alexandrou, T. W. Clyne and G. A. J. Amaratunga, Chem. Phys. Lett., 2003, 373, 52–56 CrossRef CAS.
- X. Wang, B. Xu, X. Liu, H. Jia and I. Hideki, Phys. B, 2005, 357, 277–281 CrossRef CAS.
- Q. Zou, M. Z. Wang, Y. G. Li, B. Lv and Y. C. Zhao, J. Exp. Nanosci., 2010, 5, 473–487 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Philos. Trans. R. Soc., A, 2004, 362, 2477–2512 CrossRef CAS PubMed.
- A. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS.
- C. M. Julien, M. Massot and C. Poinsignon, Spectrochim. Acta, Part A, 2004, 60, 689–700 CrossRef CAS.
- C. Julien, M. Massot, R. Baddour-Hadjean, S. Franger, S. Bach and J. Pereira-Ramos, Solid State Ionics, 2003, 159, 345–356 CrossRef CAS.
- D. Weingarth, H. Noh, A. Foelske-Schmitz, A. Wokaun and R. Kötz, Electrochim. Acta, 2013, 103, 119–124 CrossRef CAS.
- V. Khomenko, E. Raymundo-Pinero and F. Béguin, J. Power Sources, 2006, 153, 183–190 CrossRef CAS.
- M. Huang, Y. Zhang, F. Li, L. Zhang, R. S. Ruoff, Z. Wen and Q. Liu, Sci. Rep., 2014, 4, 3878 Search PubMed.
- X. Zhang, P. Yu, H. Zhang, D. Zhang, X. Sun and Y. Ma, Electrochim. Acta, 2013, 89, 523–529 CrossRef CAS.
- Y. Wang, S. F. Yu, C. Y. Sun, T. J. Zhu and H. Y. Yang, J. Mater. Chem., 2012, 22, 17584–17588 RSC.
- O. Vargas, A. Caballero, L. Hernán and J. Morales, J. Power Sources, 2011, 196, 3350–3354 CrossRef CAS.
- Z. Li, J. Wang, Z. Wang, H. Ran, Y. Li, X. Han and S. Yang, New J. Chem., 2012, 36, 1490–1495 RSC.
- X. Tang, H. Li, Z.-H. Liu, Z. Yang and Z. Wang, J. Power Sources, 2011, 196, 855–859 CrossRef CAS.
- J. Zhu and J. He, ACS Appl. Mater. Interfaces, 2012, 4, 1770–1776 CAS.
- P. Ragupathy, D. H. Park, G. Campet, H. Vasan, S.-J. Hwang, J.-H. Choy and N. Munichandraiah, J. Phys. Chem. C, 2009, 113, 6303–6309 CAS.
- S. Devaraj and N. Munichandraiah, J. Electrochem. Soc., 2007, 154, A80–A88 CrossRef CAS.
- C. Portet, G. Yushin and Y. Gogotsi, Carbon, 2007, 45, 2511–2518 CrossRef CAS.
- D. V. Ragone, SAE [Tech. Pap.], 1968, 680453 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Transmission electron micrographs of the carbon materials are shown. See DOI: 10.1039/c6ra24181f |
| This journal is © The Royal Society of Chemistry 2016 |