
Polymer supported DMAP: an easily recyclable organocatalyst for highly atom-economical Henry reaction under solvent-free conditions†

Abstract
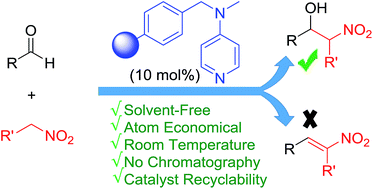
Polymer supported catalysts are regarded as a borderline class of catalysts, which retains the advantages of homogeneous catalysts while securing the ease of recovery by simple filtration and workup of heterogeneous systems. Additionally, such catalysts are less hygroscopic due to the long polymer backbone. Here we have demonstrated that a catalytic amount of polymer supported DMAP (10 mol%) can lead to excellent conversion of an equimolar mixture of aldehyde and nitroalkane exclusively into β-nitroalcohols via the Henry reaction. Unlike most of the commonly used catalysts, polymer supported DMAP can be recovered by simple filtration and reused several times, thereby reducing the operational cost. High synthetic efficiency, total atom economy, near quantitative yields, mild reaction conditions, operational simplicity, easy recovery and reusability of the catalyst, solvent-free reaction conditions and avoidance of traditional reaction workup make the protocol highly significant from Green and Sustainable Chemistry perspectives.
 Please wait while we load your content...
Please wait while we load your content...

