DOI:
10.1039/C6RA21427D
(Paper)
RSC Adv., 2016,
6, 104164-104172
Dye-doped silica nanoparticle probes for fluorescence lifetime imaging of reductive environments in living cells†
Received
26th August 2016
, Accepted 22nd October 2016
First published on 24th October 2016
Abstract
Fluorescence detection sensitivity can be drastically improved by the application of nanoparticles (NPs) because of their superior brightness compared to organic dyes. Here, using dye-doped silica NPs (SiNPs), we developed FRET-based nanoparticle probes for the detection of reductive environments in living cells. To this end, we designed three FRET acceptors based on black hole quenchers (BHQs). Their polarity was tuned by introducing hydroxyl, PEG and sulfate groups. To conjugate them to NPs, we used an original pre-functionalization approach, where the quencher was coupled by a “click” reaction to Pluronic F127 and further used for the preparation of silica NPs. This approach enabled easy preparation of silica NPs functionalized with varying amounts of quenchers by simple mixing of functionalized and parent Pluronic F127 in different mol%. The increase in the quencher concentration at the SiNPs surface produced a rapid drop in the fluorescence intensity with 80% quenching and a 2-fold drop in the emission lifetime for 16 mol% of the quenchers. Then, to obtain turn-ON sensing of reductive environments, the quenchers were coupled to the NPs through a disulfide linker using the same pre-functionalization strategy. The obtained nano-probes showed a >10-fold increase in their fluorescence in the presence of reductive agents, such as tris(2-carboxyethyl)phosphine (TCEP) and glutathione. Remarkably, BHQ quencher bearing sulfate group showed the highest turn-ON response, probably due to its superior capacity to escape from the NP surface after disulfide bond cleavage. The obtained best nanoprobe was successfully applied for detection of reductive environments inside living cells using fluorescence lifetime imaging (FLIM). This work provides insights for FRET acceptor design and its controlled grafting, which enables preparation of the first redox-sensitive silica nanoparticle probe for lifetime imaging.
Introduction
Fluorescent probes are essential tools in current biomolecular research.1–3 They enable detection of bioactive molecules and monitoring of biomolecular processes with the highest specificity and sensitivity. However, the ever present problem of fluorescent molecular probes is their brightness, due to the limited values of their extinction coefficient and quantum yields.4 Therefore, fluorescent nanoparticles have gained recent attention because they can be 10–100 fold brighter than individual dyes.5–7 So far, a variety of fluorescent NPs has been developed: quantum dots (QDs),8,9 dye-doped silica NPs (SiNPs),10–13 nanodiamonds,14 conjugated polymer NPs,15,16 dye-doped polymer NPs,7,17–19etc. Within this list, dye-loaded silica NPs present particular interest due to the high brightness and photostability they can reach, combined with the very low toxicity of many of their formulations. Moreover, their high versatility offered by the possible synthetic strategies and by the introduction of effective collective photophysical processes18,20 makes them a powerful toolbox for addressing many different imaging and sensing applications in the field of nanomedicine.
The most common mechanism in the design of fluorescent probes is Förster Resonance Energy Transfer FRET.21 It is particularly suitable for molecular probes because dyes are very small entities, which enable preparation of FRET pairs with sufficiently small inter-fluorophore distances that ensure efficient FRET.22–24 In contrast, NPs are less efficient FRET donors or acceptors because frequently their size is much larger than the Förster radius. Thus, earlier works on quantum dots showed that efficient FRET requires ≥10 FRET acceptors at the periphery of the organic shell,25,26 though there are examples for high FRET with fewer acceptors, but located just next to the inorganic core of NPs.27 Dye-doped silica NPs are particularly promising FRET donors for two reasons. At first, recent protocols that use organic surfactant Pluronic as template enable preparation of ultrasmall NPs down to 10 nm size.11 Second, at high dye loading energy hopping within the encapsulated dyes can further improve the efficiency of the energy transfer.20,28 As we have demonstrated, this process, due to the energy transfer among chromophores of the same nature (the so-called “homo-FRET”) plays in fact a prominent role in distributing the excitation energy among several donor dyes, including the closest ones to the final acceptors.20 In this way, the silica NPs can be employed as “excitation energy reservoirs”, leading to nanoarchitectures that can be advantageously designed, for example, as nanosensors for ions,29,30 pH,31 small bioactive molecules,32,33 nucleic acids,34etc. as well as photoswitchable systems,35 large Stokes shift emitters,36,37 and fluorescent drug delivery systems.38–40
Herein we describe the development of FRET-based nanosensors using rhodamine-doped silica nanoparticles (SiNPs) linked to amphiphilic BHQ-2 type quenchers through a reducible disulfide (S–S) group. Disulfide bond has been proven to be a robust cleavable bond in the reductive environment of the cells.41–43 We varied the polarity of the quenchers by adding different polar heads and studied their interaction with fluorescent SiNPs as well as their ability to quench their fluorescence by direct adsorption and by covalent linkage. These nanosensors displayed a fluorescence increase (turn-ON effect) upon reduction of the disulfide bond by tris(2-carboxyethyl)phosphine (TCEP) or glutathione that separate the BHQ quencher from the particles. Moreover, this quenching system showed significant effect on the fluorescence lifetime of the SiNPs, which allowed us to use this system in Fluorescence Lifetime Imaging Microscopy (FLIM) experiments in live cells.
Results and discussion
Design of the nanoprobes
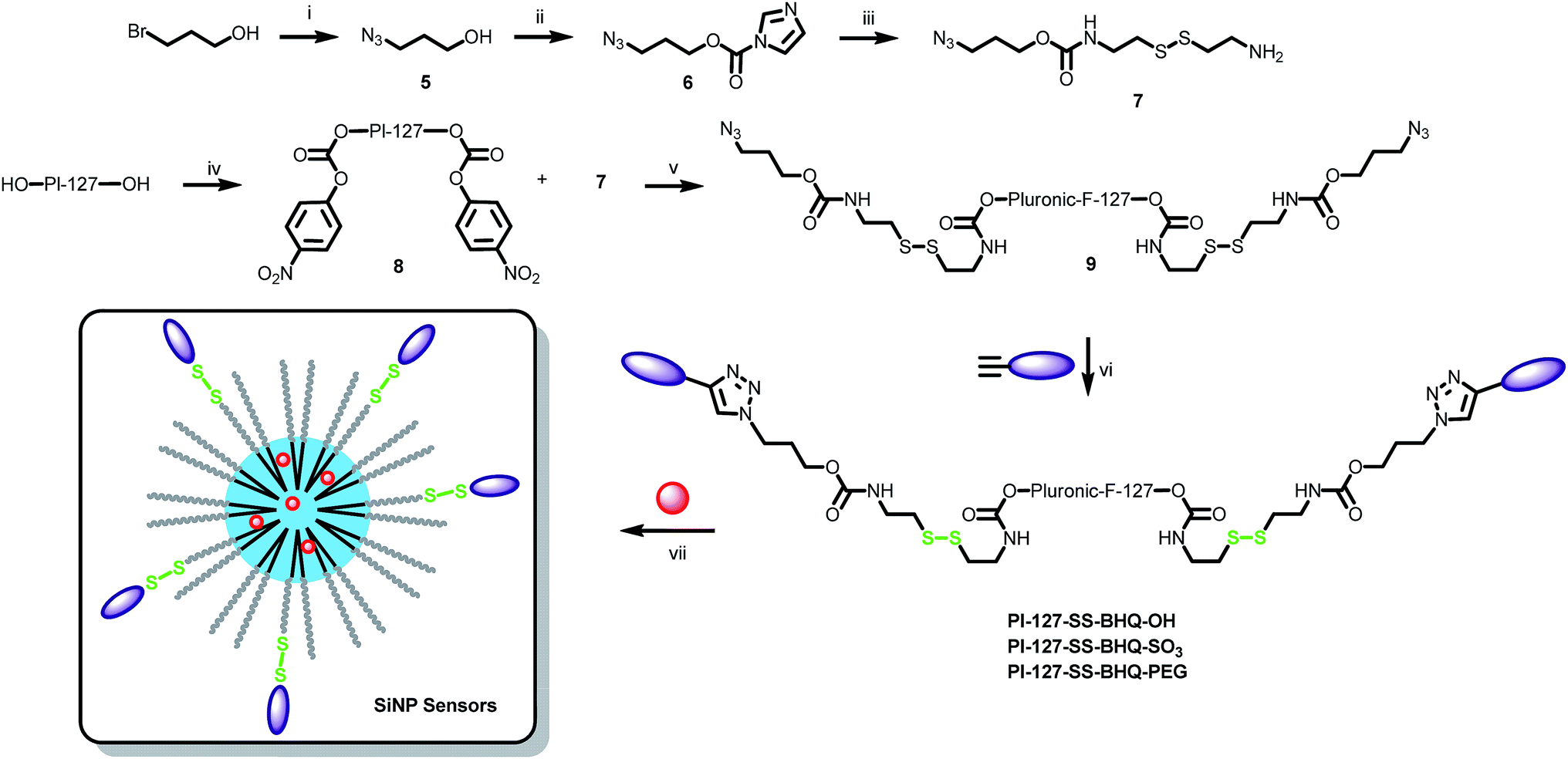
We designed a system where a fluorescent SiNP bears covalently linked quenchers through a disulfide bond. Upon reduction of this bond, the quenchers separate from the particle and provoke a fluorescence increase as the particle recovers its initial fluorescence. As a particle we used previously reported rhodamine B-doped SiNPs, prepared using Pluronic F127 as a scaffold.36 These NPs feature ∼10 nm core diameter and ∼5 nm size PEG shell. In order to achieve an efficient turn-ON of these SiNPs, the quencher must fulfil several properties. First, the overlap between the particle's emission spectrum and the absorbance spectrum of the quencher must be sufficient to insure an appropriate quenching effect. Therefore, Black Hole Quencher-2 (BHQ-2) was chosen as FRET acceptor. Then, the quencher should present an intermediate polarity, so that it could locate inside the PEG shell and thus close to the fluorescent core of the SiNPs to ensure efficient FRET. On another hand, once the disulfide bond get cleaved, the quencher moiety must be sufficiently hydrophilic so that it can escape from the particle in order to recover the fluorescence of SiNPs providing an optimal turn-ON effect. However, BHQ quenchers are non-charged apolar molecules and therefore have a lipophilic nature that could be detrimental for their escape from SiNPs. We consequently designed and synthesised three different functionalizable and amphiphilic quenchers: BHQ–OH, BHQ–SO3 and BHQ–PEG that bear, in addition to a “clickable” alkyne group, different polar heads, i.e., hydroxyl, sulphate and methoxy-PEG8 groups, respectively (Fig. 1).
 |
| | Fig. 1 Structure of the 3 amphiphilic functionalizable BHQs. Polar heads are highlighted in red, the functionalizable site is highlighted in blue. | |
At the first step of the synthesis of BHQ–OH, N-(2-hydroxyethyl)aniline was reacted with propargyl bromide to introduce a clickable group. The obtained aniline was reacted with Fast Black K giving rise to BHQ–OH (Scheme 1). BHQ–OH was efficiently converted to BHQ–SO3 by sulfation using the SO3·NEt3 complex with a yield of 87%. BHQ–OH was clicked to a methoxy PEG8 moiety to afford compound 3. The latter was then converted into an active carbonate 4, which was coupled to propargylamine to obtain BHQ–PEG (Scheme 1).
 |
| | Scheme 1 Synthesis of the 3 “clickable” BHQs: (i) propargyl bromide, NaHCO3, DMSO; (ii) Fast Black K salt, dioxane/water; (iii) SO3·NEt3, DMF; (iv) SOCl2, pyridine, CHCl3, NaN3, DMF (2 steps); (v) 2, CuSO4·5H2O, AscNa, dioxane/water; (vi) 4-nitrophenyl chloroformate, pyridine, DCM; (vii) propargylamine, DIEA, DMF. | |
Model FRET experiments
These amphiphilic quenchers were first used to study their ability to interact non-specifically with the surface of SiNPs. Increasing amounts of BHQs were added to an aqueous solution of rhodamine B doped SiNPs and the fluorescence spectra were recorded. It was found that the quenching efficiency was dependent on the structure of the quencher (Fig. 2). At a ratio of two BHQ molecules per SiNP (10 rhodamine B dyes per SiNP, calculated as previously reported36), BHQ–OH, BHQ–PEG and BHQ–SO3 quenched 30, 40 and 55% of the SiNP fluorescence, respectively. At 10 quenchers per NP, nearly 80% of the SiNP fluorescence was quenched for all three quenchers. Thus, all three quenchers displayed capacity to bind SiNPs non-specifically, though this behaviour depended slightly on the nature of the polar group.
 |
| | Fig. 2 Effect of the adsorption of the 3 BHQs on the SiNPs' fluorescence intensity (excitation wavelength = 530 nm); concentration of SiNPs = 200 nM. | |
FRET to covalently grafted quenchers
In a second step, the quenching ability of these amphiphilic BHQs, being covalently attached to the surface of SiNPs, was investigated. In one approach, rhodamine B doped SiNPs bearing an azide group at the terminal end of the Pluronic F127 (Pluronic) were prepared.44 The functionalization of the SiNPs with the clickable quenchers by CuAAC “click” chemistry was not efficient, as we could not see significant differences between the samples with and without Cu-catalyst. This could be explained by the poor efficiency of the “click” reaction at low concentrations in the presence of oxygen. Higher efficiency would require the use of a special chelated Cu(I) catalyst.45 In a second approach, Pluronic-N3 was directly functionalised with the quenchers by CuAAC click chemistry prior to the formulation of SiNPs. The reaction proceeded smoothly and provided pure bi-functionalised Pluronic F127 (Scheme 2) according to NMR. Then, the modified Pluronic polymer was used to prepare SiNPs using the same protocol. Previously, it was estimated that a single SiNP was constituted of ∼50 Pluronic polymers,46,47 which corresponded to ∼100 functionalizable sites per particle. Therefore, SiNPs bearing 0, 2, 4, 8 and 16 BHQs at their surface were readily obtained by mixing Pluronic and Pluronic–BHQ in appropriate ratios prior to formulation, i.e. 0, 2, 4, 8 and 16% of Pluronic–BHQ in total Pluronic, respectively. Dynamic light scattering (DLS) measurements showed that the Pluronic–BHQ molecules did not affect the size (24 nm) and the low polydispersity value (typically <0.1) of the obtained SiNPs (Table 1). This important result shows that Pluronic modified with quencher can be readily used as a building block to prepare functionalized SiNPs with preserved size characteristics.
 |
| | Scheme 2 Two different approaches for the preparation of functionalised SiNPs by CuAAC click chemistry. (i) MsCl, NEt3, DCM; (ii) NaN3, CH3CN. | |
Table 1 Hydrodynamic diameter and polydispersity of the different SiNPs as measured by DLS
| NPs |
BHQ (%) |
Diameter (nm) |
Polydispersity (PDI) |
| SiNPs |
0 |
22 |
0.02 |
| SiNPs–N3 |
0 |
22 |
0.1 |
| SiNPs–BHQ–OH |
16 |
23 |
0.01 |
| SiNPs–SS–BHQ–OH |
16 |
25 |
0.1 |
| SiNPs–SS–BHQ–OH |
32 |
36 |
0.28 |
| SiNPs–BHQ–PEG |
16 |
24 |
0.01 |
| SiNPs–SS–BHQ–PEG |
16 |
24 |
0.03 |
| SiNPs–SS–BHQ–PEG |
32 |
25 |
0.06 |
| SiNPs–BHQ–SO3 |
16 |
24 |
0.01 |
| SiNPs–SS–BHQ–SO3 |
16 |
29 |
0.25 |
| SiNPs–SS–BHQ–SO3 |
32 |
26 |
0.3 |
The increase in the fraction of Pluronic–BHQ during particle synthesis resulted in the gradual increase in the absorbance of the obtained SiNPs as well as broadening of the absorption band with appearance of a shoulder above 600 nm (Fig. 3A). These spectral changes correspond to the increased contribution of the absorption of the BHQ quenchers, which confirms their successful grafting to SiNPs. Fluorescence measurements of the obtained SiNPs–BHQs showed that an increase in the number of BHQ molecules per particle leads to a gradual decrease of the fluorescence intensity (Fig. 3B and C). Despite their different polar groups, the quenching efficiency was similar for all three quenchers, so that 16 BHQ molecules per particle quenched more than 80% of SiNPs fluorescence in all cases (Fig. 3C). The average fluorescence lifetime also showed a gradual decrease upon an increase in the number of BHQ quenchers (Fig. 3D), though this effect was slightly stronger for BHQ–OH. Overall, the lifetime decreased from ∼3.7 ns in the parent SiNPs up to 1.9–2.1 ns in SiNPs bearing 16 quenchers per particle. It should be noted that the decrease in the lifetime is smaller than the decrease in the fluorescence intensity, namely, <2-fold and ∼5-fold, respectively. This discrepancy indicates that FRET is probably not the only mechanism of quenching of SiNPs by BHQ. We expect that other mechanisms (electron transfer and excitonic interactions) could produce dark species that decrease the fluorescence intensity without detectable change in the lifetime measurements. Moreover, we cannot exclude a non-homogeneous distribution of dyes and/or quenchers in SiNPs, which could also affect differently fluorescence intensity and the lifetime. Clearly, these experiments show that NPs could be efficiently quenched by a relatively small number of BHQ quenchers and this process can be followed by both intensity and lifetime measurements.
 |
| | Fig. 3 Spectroscopic properties of SiNPs with grafted BHQ quenchers. Absorption spectra (A) and emission spectra (B) of SiNP–BHQ–SO3 (200 nM) with various amounts of BHQ per particle. Excitation wavelength was 530 nm. (C and D) Quenching effect of the covalently grafted BHQ quenchers on the fluorescence intensity (C) and the average lifetime (D) of SiNPs. | |
Preparation of nanoprobes and their validation in solution
Encouraged by the developed synthetic methodology and the quenching efficiency of the new amphiphilic BHQs, we prepared a fluorogenic nanoprobe. The new BHQs quenchers were attached to the terminal PEG chains of the Pluronic F127 through a linker containing a disulfide bond. For this purpose, bromopropanol was converted into alcohol azide 5, which was activated by carbonyldiimidazole 6. The latter was coupled to cystamine to give an amino-azide linker 7. Pluronic F127 was converted to a di-active carbamate 8 that was subsequently reacted with 7 to afford the Pluronic azide derivative 9. The latter was clicked to the 3 different amphiphilic BHQs (Scheme 3). The reducible Pluronic-quencher conjugates were then mixed with Pluronic F127 in ratios 4/21 and 8/17 prior to the formulation to obtain SiNPs with respectively 16 and 32 BHQs at their surface. These ratios were chosen to insure an efficient quenching and therefore an enhanced turn-ON effect upon reduction.
 |
| | Scheme 3 Synthesis of the reducible SiNPs nanosensors. (i) NaN3, DMF; (ii) carbonyldiimidazole, EtOAc. (iii) Cystamine, DCM; (iv) 4-nitrophenyl chloroformate, pyridine, DCM; (v) DIEA, CH3CN; (vi) BHQ, CuSO4·5H2O, AscNa, dioxane/water. (vii) Pluronic F127, rhodamine B–Si(OEt)3, AcOH, TEOS, TMSCl. | |
As expected, the fluorescence intensity decreased many-fold for all three quenchers and the effect was particularly strong with 32% of quenchers per particle (Fig. S1†). To evaluate the ability of these nano-constructs to sense a reductive environment, they were incubated with 20 mM TCEP (pH was adjusted to 7.4 by sodium hydroxide) or 40 mM glutathione and their fluorescence intensity was monitored over one hour. For all quenchers used at 32%, TCEP produced a strong increase in the fluorescence intensity on the time scale of 1 h (Fig. 4A). The fluorescence increase was lower for SiNPs with lower % of the quencher (16%) and no increase was observed for SiNPs without grafted quencher (Fig. 4B, S2 and S3†). Addition of glutathione also produced fluorescence increase when the quencher was grafted to SiNPs, though the fluorescence intensity increased slower, probably because glutathione is a weaker reducing agent than TCEP (Fig. S4†). Thus, for SiNPs with 32% BHQ–SO3 in presence of 40 mM glutathione, the fluorescence intensity reached a plateau only after 10 h with a 12-fold enhancement (Fig. S5†), which is close to the value obtained in the presence of TCEP in 1 hour (Fig. 4A). The fluorescence intensity of the nanosensors (with S–S bond) in the absence of reducing agent was nearly invariant for at least 20 min, which demonstrated their stability in the phosphate buffer medium (Fig. S6†). An invariant fluorescence over time was also observed for BHQ-free SiNPs in the presence of glutathione, thus, excluding a direct effect of the reducing agent on SiNPs (Fig. S7†). Finally, in our control SiNPs–BHQ–OH without reducible linker, no fluorescence increase was observed in the presence of glutathione (Fig. S8†). Taken together, these results suggest that the reducing agents cleave the S–S bonds, which leads to the liberation of the quenchers and thus to the increase in the SiNPs fluorescence.
 |
| | Fig. 4 Effect of TCEP on the fluorescence of the fluorogenic SiNPs. (A) Fluorescence increase as a function of time of BHQ–SO3 SiNPs (20 nM) in the presence of 20 mM TCEP in phosphate buffer 20 mM pH 7.4. (B) Fluorescence increase as a function of time of SiNPs bearing 32% BHQ–OH (red), BHQ–SO3 (black) and BHQ–PEG (green) (20 nM) in the presence of 20 mM TCEP in phosphate buffer 20 mM pH 7.4. Excitation and emission wavelengths were 530 nm and 580 nm, respectively. | |
The three nanosensors (at 32% of grafting) displayed significantly different turn-ON effect efficiencies upon reduction as their fluorescence enhancement varied from 3- to 13-fold after one hour in the presence of TCEP (Fig. 4A) and 1.5 to 3.5-fold after one hour with glutathione (Fig. S4†). The following ranking of BHQ efficiency in terms of turn-ON effect for the nanosensors could be drawn up: BHQ–SO3 > BHQ–OH > BHQ–PEG. This tendency can be explained by the fact that, in these systems, an efficient turn-ON effect is ensured by three synergetic factors: (1) the accessibility of the S–S bond; (2) the capacity of BHQ to quench NPs and (3) the capacity of the BHQ to escape from SiNPs after S–S bond reduction. For all three quenchers we expect that the access to S–S bond to the reductive agent should be similar. Moreover, all three quenchers at high percentage of grafting showed similar quenching efficiency. Therefore, the observed differences are probably related to the differences in desorption of the quenchers from the SiNPs after S–S bond reduction. The presence of a negative charge brought by the sulfate group of BHQ–SO3 is likely favorable to the dissociation of the BHQ from the fluorescent SiNP, that have a negative zeta potential, leading to an efficient de-quenching of this latter. By contrast, BHQ–PEG and BHQ–OH are non-charged which can explain their lower efficiency compared to BHQ–SO3 due to less efficient desorption from SiNPs after S–S bond reduction. More unexpected was the significant difference in efficiencies between BHQ–PEG and BHQ–OH. Indeed BHQ–OH was shown to be >2 times more efficient than its PEGylated version despite the fact that the latter bears two triazole moieties as well as a PEG8 which enhance its polarity.
Validation in living cells
The efficient turn-ON effect observed in the SiNP nanoprobes bearing 32% BHQ–SO3 led us to investigate their ability to sense the reductive environment in living cells. HeLa cells were incubated for 2 hours in the presence of SiNPs with no quencher and SiNPs with 32% of BHQ–SO3. In both cases the cells showed intracellular dotted fluorescence, which can be ambiguously assigned to internalized SiNPs (Fig. 5). The internalization of these nanoprobes probably took place by endocytosis, as suggested by the earlier data using analogous SiNPs.48 As SiNPs with 32% BHQ–SO3 are very poorly fluorescent in the intact form, the observation of fluorescence inside the cells comparable to that of control NPs without grafted quencher indicates that the internalized nanoprobes probably turned ON their fluorescence due to the reductive environment present in cytosol and endosomes of living cells.41,49 However, as intensity response cannot provide definite answer regarding the particle turn-ON, we performed Fluorescence Lifetime Imaging Microscopy (FLIM), which can provide direct information about changes in FRET.50,51
 |
| | Fig. 5 Confocal laser scanning microscopy images of SiNP-0% BHQ (A) and SiNP-32%–SS–BHQ–SO3 (B) incubated at 50 nM for 2 h in the presence of HeLa cells. SiNPs were excited at 561 nm and were detected in the 567–700 nm range (red color). The plasma membrane was stained by WGA-AlexaFluor®488 (green). Scale bar is 10 μm. | |
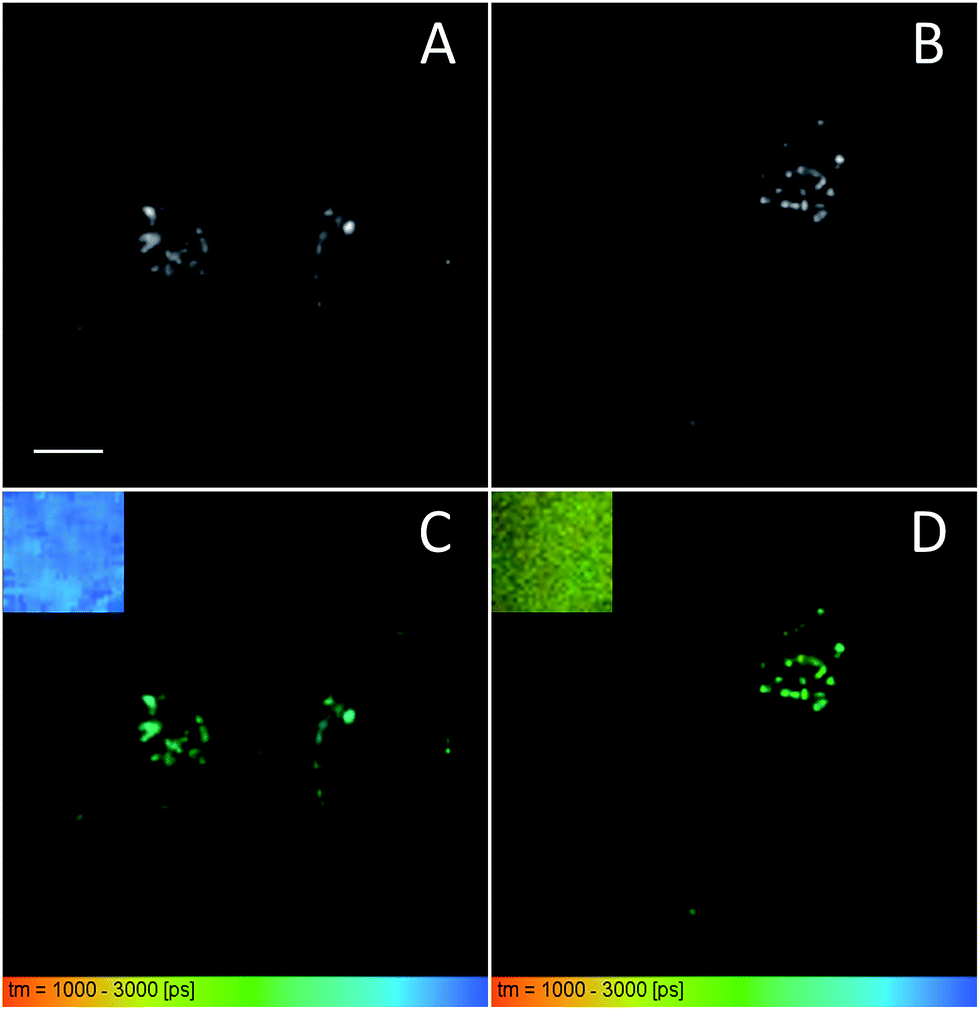
In our FLIM studies, we first compared the images of suspensions of control SiNPs and SiNPs bearing 32% of BHQ–SO3 quenchers (Fig. 6). They showed clearly different pseudo-colors (blue and yellow-green), with average lifetimes around 2.9 and 1.8 ns. Thus, in line with our time-resolved studies (see above), the BHQ-bearing SiNPs showed significantly shorter lifetime due to FRET quenching process. Then, we incubated these two types of SiNPs with HeLa cells for 2 h. The obtained FLIM images showed strong intracellular fluorescence in form of dots, indicating good internalization of both types of SiNPs (Fig. 6). However, in the case of SiNPs bearing BHQ–SO3, the pseudo-color showed clear heterogeneity (Fig. 6D), ranging between long lifetime of control SiNPs (blue) and the short lifetime of SiNPs with 32% of BHQ–SO3 (yellow-green). This result suggests that after internalization, some part of the nanoprobes underwent reductive cleavage at the S–S bond, which produced de-quenched SiNPs with lifetime close to control SiNPs. Thus, FLIM images validated the response of our nanoprobes to the reductive stimulus in the living cells. Moreover, we showed that FLIM imaging applied to FRET nanoprobes is complementary to intensity measurements, because FLIM provides information about FRET efficiency, thus describing the probe response to the reductive environment.
 |
| | Fig. 6 Intensity (A and B) and FLIM (C and D) images of SiNP-0% BHQ (A and C) and SiNP-32%–SS–BHQ–SO3 (B and D) incubated at 50 nM for 2 h in the presence of HeLa cells. Insets of (C) and (D) at the top left corner show the FLIM image of corresponding SiNPs without (SiNP-0% BHQ) and with BHQ quencher (SiNP-32%–SS–BHQ–SO3) in solution. Scale bar is 10 μm. | |
Materials and methods
Milli-Q water (Millipore) was used in all experiments. All starting materials for synthesis were purchased from Alfa Aesar, Sigma Aldrich or TCI Europe and used as received unless stated otherwise. Synthesis of the BHQ and the Pluronic F127 derivatives are described in the ESI.† NMR spectra were recorded on a Bruker Avance III 400 MHz spectrometer. Mass spectra were obtained using an Agilent Q-TOF 6520 mass spectrometer. DLS measurements were performed on a Zetasizer Nano series DTS 1060 (Malvern Instruments S.A.). Absorption and emission spectra were recorded on a Cary 400 Scan ultraviolet-visible spectrophotometer (Varian) and a FluoroMax-3 spectrofluorometer (Horiba Jobin Yvon) equipped with a thermostated cell compartment, respectively. For standard recording of fluorescence spectra, the excitation wavelength was set to 530 nm and emission was recorded from 540 to 700 nm. The solutions of TCEP and glutathione were prepared in 20 mM phosphate buffer solutions (pH 7.4) and the pH was readjusted to 7.4.
Cell culture
HeLa cells (ATCC CCL-2) were grown in DMEM (Gibco-Invitrogen), supplemented with 10% fetal bovine serum (Lonza) and 1% antibiotic solution (penicillin–streptomycin, Gibco-Invitrogen) at 37 °C in humidified atmosphere containing 5% CO2. Cells were seeded onto a chambered coverglass (IBiDi) at a density of 5 × 104 cells per well 24 h before the microscopy measurements. For imaging, the culture medium was removed and the attached cells were washed with Opti-MEM (Gibco-Invitrogen). Next, a freshly prepared solution of NPs (50 nM) in Opti-MEM was added to the cells, which were incubated for 2 or 24 h. The medium was then removed and washed 2 times with HBSS and replaced by HBSS for imaging. Cell plasma membrane staining with wheat-germ agglutinin-Alexa488 (50 nM) was done at room temperature 10 min before the measurements.
Fluorescence lifetime measurements in solution
Time-resolved fluorescence measurements were performed with the time-correlated, single-photon counting technique using the excitation pulses at 480 nm provided by a pulse-picked frequency doubled Ti–sapphire laser (Tsunami, Spectra Physics) pumped by a Millenia X laser (Spectra Physics). The emission was collected through a polarizer set at the magic angle and an 8 nm band-pass monochromator (Jobin-Yvon H10) at 582 nm. The single-photon events were detected with a micro-channel plate photomultiplier (Hamamatsu) coupled to a pulse pre-amplifier HFAC (Becker-Hickl GmbH) and recorded on a time correlated single photon counting board SPC-130 (Becker-Hickl GmbH). The instrumental response function (IRF) was recorded using a polished aluminum reflector, and its full-width at half-maximum was ∼40 ps. Experimentally measured fluorescence decays were deconvoluted with the instrumental response function and fitted to retrieve the most probable lifetime distribution using the maximum entropy method (Pulse 5 software).52,53 In all cases, the χ2 values were close to 1, indicating an optimal fit.
Fluorescence lifetime imaging microscopy
Fluorescence microscopy experiments were performed on a home-built two-photon laser scanning setup based on an Olympus IX70 inverted microscope with an Olympus 60× 1.2 NA water immersion objective.54 Two-photon excitation was provided by a Ti–sapphire (Tsunami, Spectra Physics) or an Insight DeepSee (Spectra Physics) laser. Imaging was carried out using two fast galvo-mirrors operating in the descanned fluorescence collection mode. Photons were detected with an avalanche photodiode (APD SPCM-AQR-14-FC, PerkinElmer) which was connected to a time-correlated single photon counting (TCSPC) module (SPC830, Becker and Hickl) operating in reversed start–stop mode. The typical acquisition time was 60 s with an excitation power around 5 mW (830 nm) at the laser output. Subsequent data analysis using the software SPC Image (Becker and Hickl) allowed us to extract the fluorescence lifetimes from the decays, and their visualization in a FLIM image, using an arbitrary color scale.
Conclusions
Preparation of FRET-based sensors using nanoparticles remains a challenge because particle size is generally larger than the Förster distance. The problem can be solved by using very small NPs and a large number of quencher moieties. Here, we designed a nanosensor where fluorescent dye-doped silica NPs (SiNP) bear covalently linked quenchers through a disulfide bond. We prepared ∼20 nm silica NPs doped with rhodamine B dyes and grafted with BHQ-2 type quenchers. It was found that optimal quenching (>10-fold) requires 16 to 32 quenchers at the SiNP surface. Moreover, the amphiphilic nature of the quencher was found to play a key role in the design of these nanosensors. Time-resolved fluorescence measurements suggested in addition to the expected FRET mechanism, the presence of dark species due to static quenching. In the presence of TCEP or glutathione, SiNPs strongly increase their fluorescence due to the reduction of the disulfide bonds and separation of the quenchers from the particle. Laser scanning confocal imaging showed that particles can be internalized in cells. Fluorescence lifetime measurements (FLIM) revealed populations of internalized NPs with short and long lifetimes corresponding to weakly and strongly reduced SiNPs, respectively. Our data confirmed that the SiNPs nanoprobes lose their quenchers in response to the intracellular reductive environment. This work provides insights into design of FRET-based nanoparticle probes and demonstrates suitability of FLIM for characterization of these nanoprobes in living cells.
Acknowledgements
This work was supported by the European Research Council ERC Consolidator grant BrightSens 648528, Université de Strasbourg (IdEX 2015, W15RAT68), Global Grant Spinner 2013, Emilia-Romagna region and by Marco Polo grant, University of Bologna.
References
- B. N. G. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS PubMed.
- X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590–659 CrossRef CAS PubMed.
- L. E. Santos-Figueroa, M. E. Moragues, E. Climent, A. Agostini, R. Martínez-Máñez and F. Sancenón, Chem. Soc. Rev., 2013, 42, 3489–3613 RSC.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS PubMed.
- O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC.
- P. D. Howes, R. Chandrawati and M. M. Stevens, Science, 2014, 346, 1247390 CrossRef PubMed.
- A. Reisch and A. S. Klymchenko, Small, 2016, 12, 1968–1992 CrossRef CAS PubMed.
- W. C. W. Chan and S. M. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Chem. Soc. Rev., 2014, 43, 4243–4268 RSC.
- S. Bonacchi, D. Genovese, R. Juris, M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Angew. Chem., Int. Ed., 2011, 50, 4056–4066 CrossRef CAS PubMed.
- J. E. Lee, N. Lee, T. Kim, J. Kim and T. Hyeon, Acc. Chem. Res., 2011, 44, 893–902 CrossRef CAS PubMed.
- K. M. Wang, X. X. He, X. H. Yang and H. Shi, Acc. Chem. Res., 2013, 46, 1367–1376 CrossRef CAS PubMed.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, Nat. Nanotechnol., 2012, 7, 11–23 CrossRef CAS PubMed.
- Y. Rong, C. Wu, J. Yu, X. Zhang, F. Ye, M. Zeigler, M. E. Gallina, I.-C. Wu, Y. Zhang, Y.-H. Chan, W. Sun, K. Uvdal and D. T. Chiu, ACS Nano, 2013, 7, 376–384 CrossRef CAS PubMed.
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
- K. Li and B. Liu, Chem. Soc. Rev., 2014, 43, 6570–6597 RSC.
- A. Reisch, P. Didier, L. Richert, S. Oncul, Y. Arntz, Y. Mely and A. S. Klymchenko, Nat. Commun., 2014, 5, 4089 CAS.
- K. Trofymchuk, A. Reisch, I. Shulov, Y. Mely and A. S. Klymchenko, Nanoscale, 2014, 6, 12934–12942 RSC.
- D. Genovese, E. Rampazzo, S. Bonacchi, M. Montalti, N. Zaccheroni and L. Prodi, Nanoscale, 2014, 6, 3022–3036 RSC.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4588 CrossRef CAS PubMed.
- J. L. Fan, M. M. Hu, P. Zhan and X. J. Peng, Chem. Soc. Rev., 2013, 42, 29–43 RSC.
- A. E. Albers, V. S. Okreglak and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 9640–9641 CrossRef CAS PubMed.
- G. Leriche, G. Budin, Z. Darwich, D. Weltin, Y. Mely, A. S. Klymchenko and A. Wagner, Chem. Commun., 2012, 48, 3224–3226 RSC.
- I. L. Medintz, A. R. Clapp, H. Mattoussi, E. R. Goldman, B. Fisher and J. M. Mauro, Nat. Mater., 2003, 2, 630–638 CrossRef CAS PubMed.
- A. I. Zamaleeva, M. Collot, E. Bahembera, C. Tisseyre, P. Rostaing, A. V. Yakovlev, M. Oheim, M. de Waard, J.-M. Mallet and A. Feltz, Nano Lett., 2014, 14, 2994–3001 CrossRef CAS PubMed.
- J. Valanciunaite, A. S. Klymchenko, A. Skripka, L. Richert, S. Steponkiene, G. Streckyte, Y. Mely and R. Rotomskis, RSC Adv., 2014, 4, 52270–52278 RSC.
- K. Trofymchuk, L. Prodi, A. Reisch, Y. Mély, K. Altenhöner, J. Mattay and A. S. Klymchenko, J. Phys. Chem. Lett., 2015, 6, 2259–2264 CrossRef CAS PubMed.
- M. Arca, C. Caltagirone, G. De Filippo, M. Formica, V. Fusi, L. Giorgi, V. Lippolis, L. Prodi, E. Rampazzo, M. A. Scorciapino, M. Sgarzi and N. Zaccheroni, Chem. Commun., 2014, 50, 15259–15262 RSC.
- E. Oliveira, D. Genovese, R. Juris, N. Zaccheroni, J. L. Capelo, M. M. M. Raposo, S. P. G. Costa, L. Prodi and C. Lodeiro, Inorg. Chem., 2011, 50, 8834–8849 CrossRef CAS PubMed.
- S. Kim, H. E. Pudavar and P. N. Prasad, Chem. Commun., 2006, 2071–2073 RSC.
- L. S. Khabbaz, M. Hassanzadeh-Khayyat, P. Zaree, M. Ramezani, K. Abnous and S. M. Taghdisi, Anal. Methods, 2015, 7, 8611–8616 RSC.
- S. M. Taghdisi, N. M. Danesh, H. R. Beheshti, M. Ramezani and K. Abnous, Nanoscale, 2016, 8, 3439–3446 RSC.
- F. Gao, P. Cui, X. X. Chen, Q. Q. Ye, M. G. Li and L. Wang, Analyst, 2011, 136, 3973–3980 RSC.
- D. Genovese, M. Montalti, L. Prodi, E. Rampazzo, N. Zaccheroni, O. Tosic, K. Altenhoner, F. May and J. Mattay, Chem. Commun., 2011, 47, 10975–10977 RSC.
- E. Rampazzo, S. Bonacchi, R. Juris, M. Montalti, D. Genovese, N. Zaccheroni, L. Prodi, D. C. Rambaldi, A. Zattoni and P. Reschiglian, J. Phys. Chem. B, 2010, 114, 14605–14613 CrossRef CAS PubMed.
- X. X. He, Y. S. Wang, K. M. Wang, M. Chen and S. Y. Chen, Anal. Chem., 2012, 84, 9056–9064 CAS.
- J. Wang, P. P. Gao, X. X. Yang, T. T. Wang and C. Z. Huang, J. Mater. Chem. B, 2014, 2, 4379–4386 RSC.
- X. X. He, L. Hai, J. Su, K. M. Wang and X. Wu, Nanoscale, 2011, 3, 2936–2942 RSC.
- Y. X. Tang, H. Hu, M. G. Zhang, J. B. Song, L. M. Nie, S. J. Wang, G. Niu, P. Huang, G. M. Lu and X. Y. Chen, Nanoscale, 2015, 7, 6304–6310 RSC.
- J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS PubMed.
- G. Gasparini, E.-K. Bang, G. Molinard, D. V. Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 CrossRef CAS PubMed.
- Y. Niko, Y. Arntz, Y. Mely, G.-i. Konishi and A. S. Klymchenko, Chem.–Eur. J., 2014, 20, 16473–16477 CrossRef CAS PubMed.
- L. Bromberg and M. Temchenko, Langmuir, 1999, 15, 8633–8639 CrossRef CAS.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192–3193 CrossRef CAS PubMed.
- P. K. Sharma and S. R. Bhatia, Int. J. Pharm., 2004, 278, 361–377 CrossRef CAS PubMed.
- G. Wanka, H. Hoffmann and W. Ulbricht, Macromolecules, 1994, 27, 4145–4159 CrossRef CAS.
- E. Rampazzo, R. Voltan, L. Petrizza, N. Zaccheroni, L. Prodi, F. Casciano, G. Zauli and P. Secchiero, Nanoscale, 2013, 5, 7897–7905 RSC.
- J. Yang, H. Chen, I. R. Vlahov, J.-X. Cheng and P. S. Low, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13872–13877 CrossRef CAS PubMed.
- H. Wallrabe and A. Periasamy, Curr. Opin. Biotechnol., 2005, 16, 19–27 CrossRef CAS PubMed.
- K. Suhling, P. M. W. French and D. Phillips, Photochem. Photobiol. Sci., 2005, 4, 13–22 CAS.
- A. K. Livesey and J. C. Brochon, Biophys. J., 1987, 52, 693–706 CrossRef CAS PubMed.
- J. C. Brochon, Methods Enzymol., 1994, 240, 262–311 CAS.
- J. P. Clamme, J. Azoulay and Y. Mely, Biophys. J., 2003, 84, 1960–1968 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21427d |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b and
Andrey S.
Klymchenko
b and
Andrey S.
Klymchenko