
Dual release of angiostatin and curcumin from biodegradable PLGA microspheres inhibit Lewis lung cancer in a mice model
Luguo
Sun  *b
and
*b
and
*b
and
Abstract
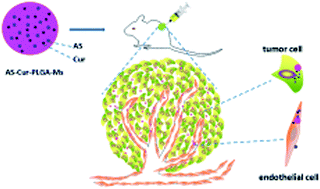
Lung cancer is an aggressive deadly disease worldwide. In this research, PLGA microspheres were used as a co-delivery system of angiostatin and curcumin for the synergistic treatment of lung cancer. The formulations were characterized by morphology, mean diameter, surface potential, and in vitro release kinetics. Furthermore, dual-drug loaded microspheres exhibited a higher antiproliferative activity in endothelial cells but not in HepG2 cells. More importantly, compared with single-drug loaded Ms (AS–PLGA-Ms and Cur–PLGA-Ms), dual-drug loaded Ms (As–Cur–PLGA-Ms) exhibited higher antitumor activity, which was further confirmed by the systemic histological and immunohistochemical analyses.
 Please wait while we load your content...
Please wait while we load your content...

