
Synthesis of α-Fe2O3, Fe3O4 and Fe2N magnetic hollow nanofibers as anode materials for Li-ion batteries
Abstract
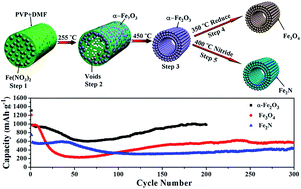
α-Fe2O3 hollow nanofibers were synthesized via a facile electrospinning process followed by a post-calcination process, and for the first time, Fe3O4 and Fe2N hollow nanofibers were successfully obtained via reduction and nitridation of the prepared α-Fe2O3 hollow nanofibers in the presence of NH3 atmosphere at 350 °C and 400 °C, respectively. The crystal structure, morphology and compositions of the α-Fe2O3, Fe3O4 and Fe2N hollow nanofibers were investigated by X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), and energy dispersive spectrometry (EDS). Electrochemical measurements show that the α-Fe2O3 and Fe3O4 hollow nanofibers electrodes deliver a high specific initial discharge capacity of 1314 and 1210 mA h g−1, respectively, and a stable cycling performance (980 mA h g−1 for α-Fe2O3 after 200 cycles and 572 mA h g−1 for Fe3O4 after 300 cycles) at a current density of 100 mA g−1. The Fe2N hollow nanofibers electrode demonstrates a high initial discharge capacity, good cycling stability (438 mA h g−1 at the 300th cycle with a current density of 100 mA g−1), high coulombic efficiency, and excellent rate capability. The superior electrochemical performances are attributed to the unique one-dimensional hollow nanostructure of the materials. The prepared hollow nanofibers are candidate anode materials for Li-ion batteries.
 Please wait while we load your content...
Please wait while we load your content...

