
An adamantane-based disubstituted binding motif with picomolar dissociation constants for cucurbit[n]urils in water and related quaternary assemblies†
E. Babjakováa,
P. Brannáa,
M. Kuczyńskaa,
M. Rouchala,
Z. Pruckováa,
L. Dastychováa,
J. Víchab and
R. Vícha*a
aDepartment of Chemistry, Faculty of Technology, Tomas Bata University in Zlín, Vavrečkova 275, 760 01 Zlín, Czech Republic. E-mail: rvicha@ft.utb.cz; Tel: +420 576 031 103
bCentre of Polymer Systems, Tomas Bata University in Zlín, třída Tomáše Bati 5678, 760 01 Zlín, Czech Republic
First published on 28th October 2016
Abstract
A non-axial centerpiece based on 1,3-disubstituted adamantane was designed, and three new guests were prepared. In the structure of the heterotritopic guests, the central adamantane site was combined with two terminal butyl or 1-adamantyl sites. The new central binding motif displayed an extraordinarily high affinity towards CB8 (Ka = (5.3 ± 0.3) × 1012 M−1 in water) to allow formation of quaternary assemblies with geometries which are dependent on the nature of macrocycles. Based on the individual binding strengths, the replacement of CB7 by CB8 led to inverse arrangements of the quaternary assemblies; i.e., β-CD is capped at the central site by two CB7 units, while the CB8 prefers the central site to be capped with two β-CD units.
Introduction
The inclusion complexes of cucurbit[n]uril (CBn) macrocycles and cationic guests that are derived from cage hydrocarbons or ferrocene have attracted significant interest in host–guest chemistry over the past two decades due to their outstanding stability.1–5 CBn is a rigid barrel-like shaped molecule with a non-polar cavity and two symmetric portals that are rimmed with carbonyl groups. Therefore, the best suited guests for CBn hosts consist of a hydrophobic central part to fill the cavity and two cationic substituents that are oriented along an axis to enable ion–dipole interactions with the opposite portals. Thus, it is not surprising that the strongest inclusion complex that has been reported to date was for 4,9-bis(trimethylammonium)-diamantane with CB7.6,7 In contrast to diamantane and other extensively used scaffolds, bicyclo[2.2.2]octane8 and ferrocene,9 which can bear two substituents that are located essentially along the axis, the two substituents at the adamantane (Ad) bridgehead positions adopt a tetrahedral orientation with an angle of 109.5°. To the best of our knowledge, there are only three examples of 1,3-disubstituted Ad dicationic guests for which the binding strengths with CB7 and CB8 have been reported.10,11 1,3-Bis(trimethyl-ammonium)adamantane diiodide has Ka values with CB7 and CB8 of 6.42 × 104 M−1 (in 50 mM CD3CO2Na/D2O) and 1.11 × 1011 M−1 (in 50 mM CD3CO2Na/D2O), respectively. Adamantane-1,3-diamine and 1,3-bis(4,5-dihydro-1H-imidazol-2-yl)adamantane have Ka values with CB7 of 2.06 × 108 M−1 (in 50 mM CD3CO2Na/D2O) and 1 × 104 M−1 (in water), respectively (the Ka values with CB8 were not reported). It should be noted that the binding strengths of these dicationic guests towards CB7 are significantly lower than those of corresponding singly substituted derivatives, where the magnitude of Ka reaches 1012 M−1.8,10 Thus, the Ad scaffold has been employed as a terminal binding site in guest molecules that display interesting supramolecular behaviors.12–17 The 1,3-disubstituted Ad cage has also been incorporated into macrocyclic molecules as a bent motif which allows for the preparation of macrobicyclic derivatives of cyclen and cyclam,18 cryptands,19 macrocyclic lactames binding squarine,20 and adamantanophanes.21,22 1,3-Disubstituted Ad has also been utilized as a suitable linker for quadruple H-bond-based binding motifs, which are capable of self-assembling into cyclopentameric complexes.23 In this paper, we present the first (to the best of our knowledge) preparation of multitopic guest molecules with a central binding site derived from a 1,3-disubstituted Ad cage (Fig. 1) and describe its binding properties. | ||
| Fig. 1 Synthesis of guests 5–7 (top) and the host molecules that were used in this study (bottom). | ||
Results and discussion
As a part of our ongoing research on multitopic guests, we prepared three new ligands (5–7; Fig. 1). Commercially available dicarboxylic acid 1 was used as the starting material for the preparation of the central motif and was converted to the compound 4 by a sequence of esterification, reduction, and the Appel reaction with a high overall yield of 71%. In the final step, we reacted 4 with three 1-alkylimidazoles to yield the corresponding bisimidazolium dibromides 5–7 at satisfactory overall yields of 43–70%. Compound 5 represents a model guest with only a central adamantane binding site, while guests 6 and 7 contain the additional terminal binding sites n-butyl and 1-adamantylmethyl, respectively, which have different affinities towards particular hosts.To overcome the steric disadvantage of 1,3-disubstituted adamantane, we used flexible ethylene linkers between the central Ad and imidazolium units. Due to the lack of information about this binding motif, we initially focused on examining the binding behavior of model guest 5 towards hosts with suitable inner cavity dimensions; i.e., CB7, CB8, and β-CD. Guest 5 interacts with β-CD following a fast exchange mode on the NMR timescale, and all of the Ad and ethylene signals are shifted downfield (see Fig. S10†). Because of the well-known magnetic anisotropy of β-CD,24 we assume that the Ad cage is included in the β-CD cavity. The association constants Ka = 1.82 × 104 M−1 (in water) and 1.71 × 104 M−1 (in 50 mM AcONa buffer), which were determined by means of isothermal titration calorimetry (ITC), are comparable to that obtained for other adamantane-based guests (Table 1).25,26 It should be noted that all binding experiments were initially performed in pure water or an aqueous NaCl solution because there is no need for buffering of the solutions of our permanent imidazolium cations. Nevertheless, we determined the thermodynamic parameters for the model guest 5 also in a sodium acetate buffer to enable easier comparison with previously published data (for full thermodynamic data, see Table S1†). By examining the complexes of 5 with CB7 and CB8 using NMR titrations, we observed rapid formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes in slow exchange mode in both cases. In addition, a significant increase of the solubility of CB8 was observed during the titration of 5 by a fine dispersion of CB8 in 50 mM NaCl. No remaining solid CB8 was observed at the final concentration of 1.1 mM. Note in Table 1 that the guest 5 is better suited for CB8 by factor of 32.3 in water (9.7 in AcONa buffer). This observation can be likely explained by better accommodation of hindered 1,3-disubstituted adamantane scaffold by wider CB8. The strength of the 5@CB7 complex (Table 1, for full ITC data, see Table S1†) is comparable to that of singly substituted adamantane-based guests. We speculate that the expected steric hindrance of the disubstituted Ad cage inside the CB7 cavity is compensated for by the ion–dipole interactions of two imidazolium rings with the CB7 portals.
:1 complexes in slow exchange mode in both cases. In addition, a significant increase of the solubility of CB8 was observed during the titration of 5 by a fine dispersion of CB8 in 50 mM NaCl. No remaining solid CB8 was observed at the final concentration of 1.1 mM. Note in Table 1 that the guest 5 is better suited for CB8 by factor of 32.3 in water (9.7 in AcONa buffer). This observation can be likely explained by better accommodation of hindered 1,3-disubstituted adamantane scaffold by wider CB8. The strength of the 5@CB7 complex (Table 1, for full ITC data, see Table S1†) is comparable to that of singly substituted adamantane-based guests. We speculate that the expected steric hindrance of the disubstituted Ad cage inside the CB7 cavity is compensated for by the ion–dipole interactions of two imidazolium rings with the CB7 portals.
| Guest | CB6 | CB7 | CB8 | β-CD |
|---|---|---|---|---|
| a All titrations were performed in triplicate. The Ka values are reported for a single binding site.b Experiments were carried out in water.c 1,6-Hexamethylene diamine·2HCl was used as a competitor.d 1-Adamantaneamine·HCl was used as a competitor.e Experiments were carried out in 50 mM AcONa.f Various fitting models were used. See Table S1 and comments in the text for detail. nb = no binding, omb = off-model binding.g Experiments were carried out in 2.5 mM NaCl. | ||||
| 5 | nb | (1.64 ± 0.09) × 1011b,c | (5.3 ± 0.3) × 1012b,d | (1.82 ± 0.01) × 104b |
| 5 | nb | (3.5 ± 0.3) × 1010c,e | (3.4 ± 0.2) × 1011d,e | (1.71 ± 0.03) × 104e |
| 6 | (1.23–1.36) × 106f,g | (1.29 ± 0.11) × 1011b,c | (3.29 ± 0.15) × 1012b,d | (1.65 ± 0.01) × 104b |
| 7 | nb | omb | omb | (0.18–6.31) × 105b,f |
To determine the possible orientation of guest 5 inside the hosts, we performed optimization calculations at the B3LYP/6-31G(d,p) level with a D3 dispersion correction and the COSMO solvent model (water) (for details and corresponding references, see Computational details). Fig. 2 shows top and side views of the minimized structures. Note that the Ad cage of 5 is shifted markedly from the virtual plane of the glycosidic oxygen atoms towards the primary rim of the β-CD likely due to steric hindrance between the C(6)H2OH groups of β-CD and the imidazolium ring. In contrast to the nearly ideal symmetric geometry of CB7 in the complex 5@CB7, with the Ad cage positioned near the center of gravity of the CB7, the Ad cage of 5 in 5@CB8 is shifted from the central position and is accompanied by tilting of the two opposite glycoluril units.
 | ||
| Fig. 2 Energy-minimized structures of the 5@β-CD (A), 5@CB7 (B), and 5@CB8 (C) complexes. | ||
Subsequently, the intrinsic stability of the 5 host aggregates in the gas phase was studied by ESI-MS. The [M + host]2+ cations were isolated in an ion trap and treated under collision-induced dissociation (CID) conditions. Fig. 3 shows the plot of the relative intensity of the [M + host]2+ signal against the CID amplitude. The observed stabilities 5@CB8 ≥ 5@CB7 > 5@β-CD correlate well qualitatively with the association constants that were obtained by calorimetric titrations, see Table 1. For comparison, we also measured 5·α-CD, which is expected to be a weak non-specific aggregate, and Ad—NH3+Cl−@CB7 with the previously reported value Ka = 4.17 × 1012 M−1.10 Although the MS data correlate well with Ka, it should be noted that fragmentation upon CID conditions combines the dissociation of the supramolecular aggregate and cleavage of its molecular components (note that Ad—NH3+ is likely much more stable than 5 in the complex with CB7).
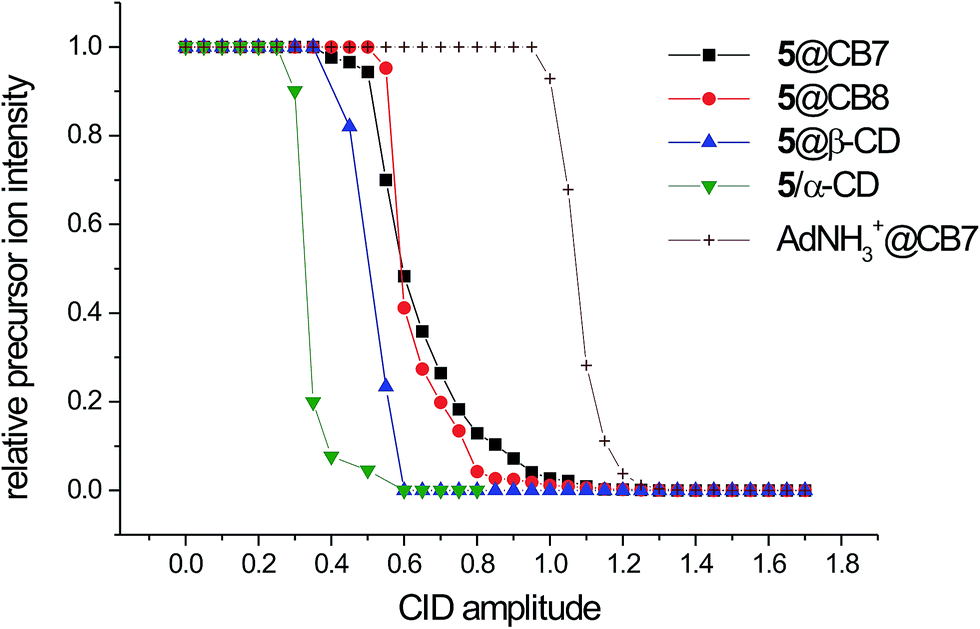 | ||
| Fig. 3 Intrinsic gas-phase stability of 5@host complexes. | ||
Having obtained these promising data, we examined the binding behavior of guest 6, which contains two additional terminal butyl binding sites. We use the superscripts “T” and “C” to denote the positioning of particular macrocycles at the terminal and central binding sites, respectively. The NMR and ITC data of the binary complexes of 6 with CB7, CB8, and β-CD indicate that the binding behavior of 6 is very similar to that of 5; i.e., 6 forms 1:1 complexes with all of these macrocycles in a pseudorotaxane manner with the central Ad cage included inside the cavity. However, in complex of 6 with CB6, hosts occupy both terminal sites with Ka values similar to those of other alkylimidazolium salts27 forming the 2:1 complex 6@CB6T2 as 1H NMR titration experiment clearly indicate (Fig. S14†). To support this hypothesis, we performed calorimetric titration of CB6 with the guest 6 which provided a single-ramp binding isotherm with inflexion at x6 = 0.47. We employed “One Set of Sites” and “Two Sets of Sites” models to obtain consistent values of Ka for single terminal site (Ka ∼ 1.3 × 106 M−1, for full data, see Table S1†). In contrast, fitting procedure using a “Sequential Binding” model did not converge on reasonable binding parameters. Upon these findings, it can be inferred that each site binds the CB6 unit independently.
Subsequently, we examined whether guest 6 is capable of forming a ternary or quaternary assembly; i.e., a complex with two different macrocycles. Our experiment can be followed by examining the stacked 1H NMR spectra in Fig. 4. Initially, four molar excess of β-CD was added to the solution of guest 6 in 50 mM NaCl in D2O (the NaCl solution was used to increase solubility of CB6). The significant downfield shift of the signals that were related to the central Ad cage implies the formation of 6@β-CDC. According to β-CD selectivity towards the butyl and the adamantane site (KAd/KBu ∼ 1000), the terminal sites remained free to allow further binding. Subsequent stepwise addition of the solution of CB6 led to a small but unambiguous downfield shift of the adamantane Hl signal. In contrast, the signals of the butyl chains were markedly shifted upfield. Broadening and/or overlapping of the peaks in the range of 1.4–1.8 ppm in final stages of titration can be attributed to increase in number of the adamantane peaks (particularly Hm and Hk) as these H-atoms became non-equivalent inside the chiral CD cavity. Considering that CB6 does not bind the Ad site due to incompatible geometries, β-CD strongly prefers the Ad site, and there is no expectation of significant repulsion between the β-CD and CB6 units, the above mentioned observations can be explained by the positioning of the two CB6 units at butyl chains, whereas β-CD remained bound to the central Ad site to form quaternary assembly 6@(β-CDC,CB6T2).
 | ||
| Fig. 4 Stacking plot of a portion of the 1H NMR (500 MHz) spectra of 6 (1.83 mM) and its complexes recorded in a 50 mM NaCl solution in D2O at 303 K. | ||
The second examined multiple-binding-site guest 7 consists of two high-affinity terminal Ad-based sites in addition to the title central site. Initially, its complexation by CB7 was studied. The NMR data (upfield shift of the terminal adamantane signals) clearly imply that the terminal Ad sites of 7 are occupied by CB7 to form 7@CB7T at low CB7 concentrations and 7@CB7T2 with excess of CB7 (see Fig. S18†). Simultaneously, the signals of the central adamantane hydrogen atoms were more shielded as the central adamantane cage was positioned close to the CB7 portals. Because no change of the guest signals intensities and/or positions was observed after addition of more than two equivalents of CB7, we infer that 7@CB7T2 predominated in the solution and no significant amount of 7@(CB7T2,CB7C) and/or 7@(CB7T,CB7C) was formed. It should be noted that formation of the two last mentioned complexes is most unlikely because such complexes, with two CB units which are arranged around one cationic moiety, suffer from strong electrostatic repulsion between two adjacent CB portals. Unfortunately, determination of the association constant for terminal Ad site of the guest 7 with CB7 via ITC was disabled because of too long equilibration when competitor was used. Although the binding constant remained unavailable, the binding isotherm which was obtained without any competitor (see Fig. 5, left) suggests that two binding sites were occupied within a simple binding event (only one slope was observed with inflexion at xCB7 ≈ 2). Combining this observation with 1H NMR data, we speculate that the ternary complex 7@CB7T2 is formed with binding strength of CB7 at AdT significantly exceeding that at AdC. If the value of Ka for AdT site and CB7 would be lower than that for AdC, the occupation of the central Ad site, not the terminal one, could be expected at low CB7 concentrations. The formation of 7@CB7T2 would follow only with the excess CB7. This is however not the case. Thus, we assume that the binding constant of CB7 at AdT is likely similar to that obtained for the model guest 1-(1-adamantylmethyl)-3-methylimidazolium iodide (AMI) and CB7 (i.e., 3.7 × 1012 M−1, for more information, see ref. 28). This would provide the two sequential bindings of CB7 at terminal Ad sites of the guest 7 with no substantial participation of the central site (Ka for AdC site of 7 could be similar to that of 5, i.e., (1.64 ± 0.09) × 1011 M−1) as depicted in Fig. 5 (left). Subsequently, we treated the guest 7 with CB8. Unfortunately, limited solubility of CB8 disabled NMR data of sufficient quality. Although the appearing of the second set of signals of aromatic hydrogen atoms clearly indicated some binding in slow-exchange manner, the signals of adamantane hydrogen atoms became too broad to allow unambiguous assignment. Binding isotherm which was recorded for competitive calorimetric titration of CB8 with the guest 7 did not fit any available model. However, two slopes were clearly observed when titration was performed without competitor as can be seen in Fig. 5 (right). Considering binding strengths of individual sites which were obtained using model compound 5 and AMI (Ka for CB8 and AMI in water at 303 K is (1.07 ± 0.15) × 1011 M−1), we assume that two subsequent distinct binding events took place when molar fraction of the guest was lower than 1.0. In other words, the CB8 unit was bound initially at AdT and then moved to AdC to form the complex 7@CB8C predominantly. This complex was transformed with the excess CB8 to 7@CB8T2 since the positioning of the two CB8 units around one cationic moiety is not preferred (Fig. 5, right). Consistent results were obtained when titrations were performed in inverse mode, i.e., the host in the cell was titrated with CBn (see Fig. S23†). These observations indicate different preferences of CB7 and CB8 towards available binding sites of the guest 7 to enable preparation of quaternary complexes with various arrangements of the CDs and CBs macrocycles as demonstrated bellow. Finally, we examined binding behavior of 7 towards β-CD. In the 1H NMR spectra that were recorded during the titration of 7 with β-CD, significant deshielding of both the central and terminal Ad cages was observed. The Job plot that was constructed for this system suggested a stoichiometry 1:2 (7:β-CD). However, the analysis of the ITC data implies that more than two binding sites can be occupied by β-CD at once (Fig. S17†).
 | ||
| Fig. 5 Assumed binding events and binding isotherms obtained by ITC for titration of the guest 7 with CB7 (left) and with CB8 (right) (guest in the cell, host in the syringe) in water at 303 K. Unlike conversions are denoted by dotted arrows. Initial concentrations: cCB7 = 0.3465 mM, c7 = 0.0036 mM; cCB8 = 0.1082 mM, c7 = 0.0084 mM. Energy differences between the two distinct 1:1 complexes were calculated using model guests. | ||
Our previous work showed that similar guests with a biphenyl central site can form rotaxane-like complexes in which one β-CD unit is firmly trapped at the central site by two CB7 units at the terminal Ad sites.28 Fig. 6 shows the 1H NMR titration experiment that confirms the ability of 7 to form a similar quaternary assembly. Initially, we added 5 eq. of β-CD to the solution of 7 in water. As discussed above, the significant downfield shift of all of the Ad signals suggests that at least two binding sites are occupied. The subsequent addition of the CB7 solution led to the large upfield shift of the terminal Ad signals, whereas the H-atoms in the central Ad cage were significantly deshielded. Because the deshielding of the central part of the guest is much higher than the deshielding that would originate solely from the portal effect of the CB7 units,27,29 we can conclude that this NMR experiment confirms the formation of the 7@(β-CDC,CB7T2) assembly. As was demonstrated in our previous work, binding of CB7 units at both terminal sites of the tritopic Ad-terminated bisimidazolium guest disabled the central biphenyl site for β-CD.28 In the case of compound 7, we observed significant deshielding of the hydrogen atoms of the central Ad cage after addition of an excess of β-CD into a solution which contained 7@CB7T2. However, we observed only single set of the β-CD signals. These observations imply that some non-specific aggregate (7@CB7T2)·β-CD is formed more likely than 7@(CB7T2,β-CDC).
 | ||
| Fig. 6 Stacking plot of a portion of the 1H NMR (500 MHz) spectra of guest 7 (1.83 mM) titrated with β-CD and CB7 in D2O at 303 K. Signals of the central Ad deshielded by β-CD are shown with asterisks. | ||
The binding data of 5 suggests a high affinity of the central site in guest 7 towards CB8. In addition, as discussed above, CB8 slightly prefers the central site over the terminal site. Subsequently, we performed 1H NMR titrations to determine whether guest 7 can form quaternary assemblies with the CB8 unit that is trapped at the central site. Although guest 7 interacts with CB8 in water, the broadening of the shielded proton signals does not allow unambiguous assignment of the signals (Fig. 7). It should be noted that during this stage of the experiment, some CB8 remained undissolved. After the subsequent addition of an excess of β-CD, the sharp signals of the central Ad appeared. Compared to the spectrum of the free guest, the signals of the central Ad are shielded, whereas the terminal Ad signals are deshielded. These observations can be rationalized by the predominant formation of complex 7@(β-CDT2,CB8C). In other words, the CB8 unit is fixed at the central binding site with two β-CDs that are positioned at the terminal sites. The molar fraction of the CB8-complexed guest was calculated immediately after the addition of β-CD using signal b (overlapping signals of all of the terminal Ad; Fig. 7) and signals i–m (related to the central Ad that was complexed with CB8) to be 0.51. However, the fraction of this quaternary assembly increased to 0.84 after 85 days of standing at room temperature. This increase can be attributed to the CB8 slow dissolving rather than slow equilibration process because of only one set of signals for CB8 was observed in the 1H NMR spectra. These CB8 signals displayed the same diffusion coefficient in a DOSY experiment as signals of the guest (see Fig. S20a in the ESI†). Additional support for the complex hypothesized above can clearly be observed in a ROESY spectrum (Fig. S19†). Whereas the cross-peaks of the terminal Ad and inner β-CD H3 and H5 suggest the inclusion of terminal Ads into β-CD cavities, the positioning of the CB8 at the central Ad can be demonstrated by the intermolecular interaction of the Hα from CB8 and Hc from the terminal adamantanes.
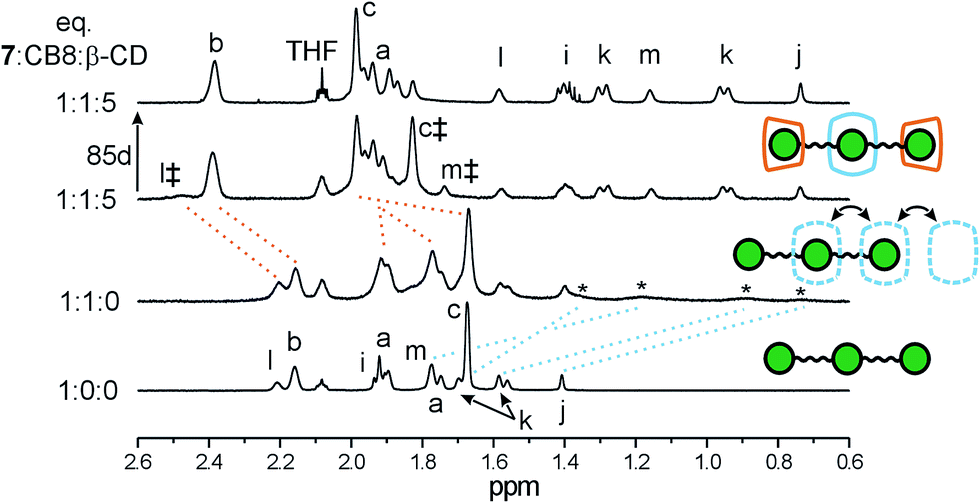 | ||
| Fig. 7 Stacking plot of a portion of the 1H NMR (500 MHz) spectra of guest 7 (1.83 mM) titrated with β-CD and CB8 in 50 mM NaCl solution in D2O at 323 K. The signals of 7@CB8 and 7@β-CD2 are denoted by * and ‡, respectively. | ||
Both quaternary assemblies consisting of guest 7 (i.e., 7@(β-CDT2,CB8C) and 7@(β-CDC,CB7T2)) were detected by ESI-MS (for the spectra, see Fig. S38 and S37,† respectively). The proposed composition of these complexes was supported by a tandem mass spectra analysis that revealed subsequent releases of the host molecules.
Experimental
General
Unless otherwise stated, all of the starting material, reagents and solvents were purchased from commercial sources and used without further purification. The adamantane-1,3-diacetic acid was obtained as a gift from Provisco CS Ltd. The melting points were measured using a Kofler block and are uncorrected. The elemental analyses (C, H, N) were performed on a Thermo Fisher Scientific Flash EA 1112. The NMR spectra were recorded on Bruker 300/500 spectrometers that operate at frequencies of 300.13/500.11 MHz (1H) and 75.77/125.77 MHz (13C). 1H and 13C-NMR chemical shifts were referenced to the solvent signals [1H: δ(residual DMSO-d5) = 2.50 ppm, δ(HDO) = 4.70 ppm, δ(residual CHCl3) = 7.26 ppm; 13C: δ(DMSO-d6) = 39.52 ppm, δ(CDCl3) = 77.16 ppm]. The signals were assigned as follows: s = singlet, t = triplet, and m = multiplet with J values in Hz. The IR spectra were recorded using KBr discs with a Mattson 3000 FT-IR instrument, and ν was reported in cm−1. The electrospray mass spectra (ESI-MS) were recorded using an amaZon X ion-trap mass spectrometer (Bruker Daltonics, Bremen, Germany) that was equipped with an electrospray ion source. All of the experiments were conducted in the positive-ion polarity mode. The instrumental conditions that were used to measure the single imidazolium salts and their mixtures with the host molecules were different; therefore, they are described separately. Single imidazolium salts: Individual samples (with a concentration of 0.5 μg cm−3) were infused into the ESI source in methanol:water (1:1, v/v) solutions using a syringe pump with a constant flow rate of 4 μl min−1. The other instrumental conditions were as follows: an electrospray voltage of −4.2 kV, capillary exit voltage of 140 V, drying gas temperature of 220 °C, drying gas flow rate of 6.0 dm3 min−1 and nebulizer pressure of 55.16 kPa. Host–guest complexes: An aqueous solution of the guest molecule (6.25 μM) and the corresponding host molecule (1.0–5.0 equivalents) for the binary/ternary complexes or an aqueous solution of the guest molecule (6.25 μM) and the corresponding host molecules (1.0–5.0 equivalents) for the quaternary complexes was infused into the ESI source at a constant flow rate of 4 μl min−1. The other instrumental conditions were as follows: an electrospray voltage of −4.0 kV, capillary exit voltage of from −30 to 140 V, drying gas temperature of 300 °C, drying gas flow rate of 6.0 dm3 min−1 and nebulizer pressure of 206.84 kPa. Nitrogen was used as the nebulizing and drying gases in all of the experiments. The tandem mass spectra were collected using collision-induced dissociation (CID) with He as the collision gas after isolating the required ions. The isothermal titration calorimetry measurements were carried out in H2O using a VP-ITC MicroCal instrument at 303 K. The concentrations of the host in the cell and the guest in the microsyringe were approximately 0.35–0.05 and 3.50–0.50 mM, respectively. The exact concentrations of CBn stock solutions were determined by the ITC titration of stable and non-hygroscopic guests which are known to form 1:1 complex (i.e., 1-adamantaneamine·HCl for β-CD, CB7, and CB8 and 1,6-hexanediamine·2HCl for CB6; purity was checked by elemental analysis prior titration). The raw experimental data were analyzed with the MicroCal ORIGIN software. The heats of dilution of each guest compound were taken into account. The data were fitted to a theoretical titration curve using the “One Set of Sites”, “Two Sets of Sites”, or “Sequential Binding” model. 1,6-Hexanediamine·2HCl with Ka,CB7 = (2.05 ± 0.09) × 109 M−1 (water) and Ka,CB7 = (2.84 ± 0.22) × 107 M−1 (50 mM CH3CO2Na buffer) and 1-adamantaneamine·HCl with Ka,CB8 = (2.79 ± 0.15) × 109 M−1 and Ka,CB8 = (2.50 ± 0.16) × 108 M−1 (50 mM CH3CO2Na buffer) were used as competitors.9 The enthalpy for competitive experiment was calculated as a sum of enthalpies for each step in competitive sequence. These ΔH values were verified by independent titration without competitor (see, Table S1†).
General procedure for the preparation of guests 5–7
The corresponding 1-methyl, 1-butyl, and 1-(1-adamantylmethyl)imidazole (4 eq.) was dissolved in dry toluene (2 cm3), and 1 eq. of 4 was added at room temperature. The mixture was stirred under inert atmosphere at 80–100 °C and monitored by thin-layer chromatography. When the starting material had been consumed, the crude product was isolated from the reaction mixture by addition of freshly distilled DEE or THF. The crude material was washed with plenty of DEE or THF under sonication and separated by centrifugation if applicable. The resultant colorless microcrystalline powder (7) or oil (5 and 6) was dried in vacuum to a constant weight; however, the elemental analyses revealed that some water and/or THF was still present. This material was used for further binding studies, and the water and/or THF molecules were taken into account.Computational details
Structures of 5, β-CD, CB7 and CB8 and corresponding inclusion complexes were optimized using B3LYP functional31,32 and standard 6-31G(d,p) basis set with COSMO (COnductor-like Screening MOdel) solvent model33 (water). The D3 dispersion correction34 was implemented during the optimizations of inclusion complexes for better description of these non-covalently bonded species.Conclusions
In conclusion, we demonstrated that the 1,3-disubstituted adamantane scaffold that bears two imidazolium cations bonded via ethylene linkers represents an efficient binding site for β-CD, CB7, and CB8. The aggregation constants of guest 5 towards β-CD and CB7 were comparable with that obtained for singly substituted adamantane-derived guests. In addition, 5 is the non-peptide guest with the highest value of Ka observed to date ((5.3 ± 0.3) × 1012 M−1 in water and (3.4 ± 0.2) × 1011 M−1 in 50 mM CH3CO2Na buffer) for 1:1 complex with CB8. Furthermore, ligands 6 and 7, which feature additional binding sites, were found to be capable of forming quaternary assemblies in a pseudorotaxane manner in a water environment. Guests 6 and 7 formed complexes with the β-CD unit at the central site capped by CB7 or CB6 at both terminal sites; i.e., 6@(β-CDC,CB6T2) and 7@(β-CDC,CB7T2). In contrast, the CB8 was predominantly trapped at the central Ad site by two β-CD units at the terminal sites; i.e., 7@(β-CDT2,CB8C). These findings can enable new modalities in the design and construction of supramolecular systems.
Acknowledgements
This work was financially supported by the Internal Founding Agency of Tomas Bata University in Zlín, project No. IGA/FT/2016/001, and the Czech Science Foundation grant 16-05691S to JV. The authors thank to Lukáš Maier from Masaryk University (Brno, Czech Republic) for assistance with the NMR measurements.Notes and references
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- D. Shetty, J. K. Khedkar, K. M. Park and K. Kim, Chem. Soc. Rev., 2015, 44, 8747–8761 RSC.
- A. E. Kaifer, Acc. Chem. Res., 2014, 47, 2160–2167 CrossRef CAS PubMed.
- L. Isaacs, Acc. Chem. Res., 2014, 47, 2052–2062 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC.
- L. Cao, M. Šekutor, P. Y. Zavalij, K. Mlinarić-Majerski, R. Glaser and L. Isaacs, Angew. Chem., Int. Ed., 2014, 53, 988–993 CrossRef CAS PubMed.
- M. Šekutor, K. Molčanov, L. Cao, L. Isaacs, R. Glaser and K. Mlinarić-Majerski, Eur. J. Org. Chem., 2014, 2533–2542 CrossRef.
- S. Moghaddam, C. Yang, M. Rekharsky, Y. H. Ko, K. Kim, Y. Inoue and M. K. Gilson, J. Am. Chem. Soc., 2011, 133, 3570–3581 CrossRef CAS PubMed.
- M. V. Rekharsky, T. Mori, C. Yang, H. K. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. Kim and Y. Inoue, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20737–20742 CrossRef CAS PubMed.
- S. Liu, C. Ruspic, P. Mukhopadhyay, S. Chakrabarti, P. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127, 15959–15967 CrossRef CAS PubMed.
- D. S. N. Hettiarachchi and D. H. Macartney, Can. J. Chem., 2006, 84, 905–914 CrossRef CAS.
- J. Gravel, J. Kempf and A. Schmitzer, Chem.–Eur. J., 2015, 21, 1–8 CrossRef PubMed.
- P. Mukhopadhyay, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2006, 128, 14093–14102 CrossRef CAS PubMed.
- X. Lu and E. Masson, Langmuir, 2011, 27, 3051–3058 CrossRef CAS PubMed.
- M. H. Tootoonchi, S. Yi and A. E. Kaifer, J. Am. Chem. Soc., 2013, 135, 10804–10809 CrossRef CAS PubMed.
- Z.-J. Ding, H.-Y. Zhang, L.-H. Wang, F. Ding and Y. Liu, Org. Lett., 2011, 13, 856–859 CrossRef CAS PubMed.
- S. Ghosh and L. Isaacs, J. Am. Chem. Soc., 2010, 132, 4445–4454 CrossRef CAS PubMed.
- S. M. Kobelev, A. D. Averin, A. K. Buryak, E. N. Savelyev, B. S. Orlinson, G. M. Butov, I. A. Novakov, F. Denat, R. Guilard and I. P. Beletskaya, ARKIVOC, 2012, 196–209 CAS.
- T. Š. Ramljak, I. Despotović, B. Bertoša and K. Mlinarić-Majerski, Tetrahedron, 2013, 69, 10610–10620 CrossRef.
- C. G. Collins, A. T. Johnson, R. D. Connell, S. A. Nelson, I. Murgu, A. G. Oliver and B. D. Smith, New J. Chem., 2014, 38, 3992–3998 RSC.
- K. Mlinarić-Majerski, D. Pavlović and Ž. Marinić, Tetrahedron Lett., 1996, 37, 4829–4832 CrossRef.
- K. Mlinarić-Majerski, D. Pavlović, M. Luić and B. Kojić-Prodić, Chem. Ber., 1994, 127, 1327–1329 CrossRef.
- H. M. Keizer, J. J. Gonzáles, M. Segura, P. Prados, R. P. Sijbesma, E. W. Meijer and J. de Mendoza, Chem.–Eur. J., 2005, 11, 4602–4608 CrossRef CAS PubMed.
- H.-J. Schneider, F. Hacket and V. Rüdiger, Chem. Rev., 1998, 98, 1755–1785 CrossRef CAS PubMed.
- M. V. Rekharsky and Y. Inoue, Chem Rev., 1998, 98, 1875–1917 CrossRef CAS PubMed.
- S. G. Kulkarni, Z. Prucková, M. Rouchal, L. Dastychová and R. Vícha, J. Inclusion Phenom. Macrocyclic Chem., 2016, 84, 11–20 CrossRef CAS.
- N. Zhao, L. Liu, F. Biedermann and O. A. Scherman, Chem.–Asian J., 2010, 5, 530–537 CrossRef CAS PubMed.
- P. Branná, J. Černochová, M. Rouchal, M. Babinský, R. Marek, M. Nečas, I. Kuřitka and R. Vícha, J. Org. Chem., 2016, 81, 9595–9604 CrossRef PubMed.
- P. Branná, M. Rouchal, Z. Prucková, L. Dastychová, R. Lenobel, T. Pospíšil, K. Maláč and R. Vícha, Chem.–Eur. J., 2015, 21, 11712–11718 CrossRef PubMed.
- J. G. Henkel, J. T. Hane and G. Gianutsos, J. Med. Chem., 1982, 25, 51–56 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. Klamt and G. Schürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- S. J. Grimme, Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra, titration data, and mass spectra. See DOI: 10.1039/c6ra23524g |
| This journal is © The Royal Society of Chemistry 2016 |