
Enhanced dielectric and energy storage performance of surface treated gallium ferrite/polyvinylidene fluoride nanocomposites†
Abstract
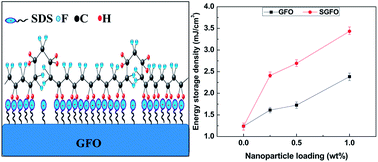
The ceramic–polymer nanocomposites composed of gallium ferrite (GFO) nanoparticles and employing sodium dodecylsulphate (SDS) as surfactant and polyvinylidene fluoride (PVDF) as matrix have been fabricated by solvent casting followed by hot-press technique. It was found that modified GFO nanoparticles favours nucleation and stabilization up to ∼84% electroactive phase (β- and γ-phase) in PVDF. Fourier transform infrared spectroscopy (FTIR) results revealed that the interfacial electrostatic interaction between nanoparticle surface charge and CH2/CF2 – molecular dipole of PVDF favoured nucleation of electroactive phase. Compared to the pristine PVDF, much higher dielectric constant (εr ∼ 25 at 10 kHz frequency) with low loss factor (tan δ ∼ 0.02 at 10 kHz) was achieved in the composite film. In addition, the nanocomposite showed higher electrical energy density (Ud ∼ 3.88 mJ cm−3 at an electric field 6 kV mm−1) compared to pristine PVDF which determined their applicability as flexible energy density capacitor.
 Please wait while we load your content...
Please wait while we load your content...

