DOI:
10.1039/C6RA23471B
(Paper)
RSC Adv., 2016,
6, 107433-107441
In situ growth of small Au nanoparticles on ZnO nanorods via ultrasonic irradiation toward super-enhanced catalytic activity†
Received
21st September 2016
, Accepted 27th October 2016
First published on 27th October 2016
Abstract
The growth process of heterostructures affects their interface feature, which governs the property. In this study, small Au nanoparticles (NPs) were in situ grown on ZnO nanorods (NRs) via a sonochemical method without adding an external reducing agent, including H atoms and free electrons, because of the reducing species generated during sonication. Because of the close attachment of the AuNPs, the Au/ZnO heterostructures exhibit superior photocatalytic performance in degradation of Rhodamine B (RhB). Namely, the Au/ZnO NRs degrade RhB completely within 12 min. It is found that the improved photocatalytic activity mainly originates from the enhanced separation of electron–hole pairs because the Au NPs act as electron traps. Moreover, the Au/ZnO photocatalyst can be reused more than five times without significant deactivation. The study of CO oxidation demonstrates that the Au/ZnO catalysts display enhanced catalytic activity, which was ascribed to the deposited Au NPs providing more active sites. It was also found that the sonochemical method is applicable for the preparation of high quality Pt/ZnO heterostructures.
1. Introduction
Photocatalysis and catalysis have attracted considerable attention due to energy and environmental problems. In particular, the photodegradation of organic pollutants and CO catalytic oxidation have become important research topics. It is critical to find effective catalytic materials to govern environmental issues, which are closely related to our daily life.1–3 Sufficient literature has demonstrated that supported Au and Pt catalysts present a unique catalytic activity for numerous vital chemical reactions.4 Noble metal nanoparticles (NPs) supported nano heterostructures have become crucial in catalysis. For various oxide-supported noble metal catalysts, some “active” supports such as CeO2 had been extensively studied and display excellent performance in the oxidation of CO.5,6 The catalytic process was usually ascribed to the Bond and Thompson mechanism,7 in which the lattice oxygen of the support can participate in the CO oxidation reaction.8 Nevertheless, differing from these “active” supports, it is unlikely for some “inert” oxides such as ZnO, TiO2 and Al2O3 to react with CO. Therefore, researchers have made considerable effort to analyze the catalytic process. For instance, Tauster and co-workers discovered the classical strong metal-support interaction (SMSI) effect in the TiO2 supported group VIII metals.9 Liu and co-workers confirmed the existence of the SMSI effect in a ZnO NR supported Au catalyst and provided a new insight into the catalytic process of “insert” oxides-novel metals in CO oxidation.1 The SMSI effect of heterostructures depends strongly on their microstructure, for example, the size distribution of noble metal NPs and the interface situation between decoration and matrix. Thus, the controlled synthesis of heterostructures is used for catalysts with high performance because the formation process determines the microstructure of the heterojunction.
One-dimensional (1D) semiconductor materials have received a lot of attention over the past few decades owing to their excellent properties such as high charge carrier mobility and catalytic efficiency, which give rise to various applications in catalysis, photoelectrochemical cells and photodetection.10,11 Among the semiconducting materials so far, ZnO is one of the most extensively investigated semiconductors because it has the advantages of being environmentally friendly, controllable morphology, easy synthesis, inexpensive and non-toxic.12,13 It has been reported that ZnO with various morphologies has diverse applications such as photocatalysis and UV photodetection.14,15 Typically, ZnO nanorods (NRs) have received considerable research interest for their applications in governing the deteriorating environment, due to their high surface-to-volume ratios, large specific surface area and efficient carrier separation, and transport performance.10 However, the major issues that limit the use of single-component ZnO NRs in the field of catalysis are the rapid recombination of electron–hole pairs generated by the absorption of photons and low photostability on account of photochemical corrosion.16,17 Hence, considerable efforts have been dedicated to improve the catalytic efficiency of ZnO NRs using diverse strategies. For example, metal NPs, metal oxide with a narrow band gap, and carbon materials are usually used to decorate the ZnO NRs to improve their charge separation.18–21 Sonochemistry is used for the synthesis of heterostructures of noble metals and ZnO NRs because of the formation of free electrons and H atoms during sonication, which can induce the noble metal ions to start the deposition in situ.22–27 The sonochemical synthesis of Au NPs gives a narrow size distribution and facile control over their morphology.22–26 Kim and co-workers successfully prepared a Au/ZnO heterostructure via a sonochemical method in an ethanol and H2O solution.27 However, the deposition of Au NPs was not homogeneous, in which Au NPs were not attached tightly on the NRs and revealed a broad size distribution. Therefore, a sonochemical method without any hole scavengers was applied in our work to slow down the rate of nucleation and growth of the Au NPs.
In this study, the Au/ZnO catalysts were successfully synthesized under ultrasonic irradiation employing ZnO NRs prepared via a hydrothermal method as the support. The reduction of Au3+ was mainly ascribed to the formation of reducing species such as H atoms and free electrons in the ultrasonic system. To test the photocatalytic performance of Au/ZnO, the as-prepared Au/ZnO samples were evaluated by the degradation of Rhodamine B (RhB) under a UV light irradiation. The enhanced photocatalytic mechanism was also discussed. The RhB degradation efficiency of the Au/ZnO NRs prepared by ultrasonication was about 4 times higher than that of the sample obtained by chemical deposition with a Au to ZnO molar ratio of 4%. In particular, the photodegradation of RhB was carried out within 12 min for Au/ZnO NRs obtained under ultrasonic irradiation. The synthesis of Pt/ZnO composites has also been realized by employing a similar sonochemical method. The Pt/ZnO catalyst also showed enhanced catalytic activity for CO oxidation. Namely, the temperature of CO conversion could be reduced to 176 °C in the case of a Pt loading of 3.2%, in which the temperature is 103 °C, which is low when compared with the pure ZnO NR samples.
2. Experimental
2.1 Chemical materials
Zinc acetate dihydrate (Zn(Ac)2·2H2O), sodium hydroxide (NaOH), sodium borohydride (NaBH4), RhB, HAuCl4·3H2O and H2PtCl6·6H2O were purchased from Shanghai Chemical Reagent Company. Anhydrous ethanol (C2H5OH) was purchased from Tianjin Fuyu Chemical Industry Co. Ltd. (Tianjin, China). All chemicals were of analytical grade and used as received without further purification. Deionized water (H2O, resistivity ∼18 MΩ cm) was obtained from a Milli-Q synthesis system.
2.2 Synthesis of the ZnO NRs
ZnO NRs were synthesized using a solvothermal method. Zn(Ac)2·2H2O (0.2195 g) and flake-like NaOH (0.4 g) in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 were dissolved in anhydrous ethanol (30 mL) with vigorous stirring for 30 min to obtain a homogeneous white solution. Subsequently, the resulting solution was transferred to a 50 mL Teflon-lined stainless autoclave and maintained at 150 °C for 24 h in an oven. After cooling down to room temperature, the white precipitates were thoroughly centrifuged and washed three times with H2O and C2H5OH. Finally, the ZnO NRs were collected for further experiments and characterization after drying at 60 °C for 10 h.
:10 were dissolved in anhydrous ethanol (30 mL) with vigorous stirring for 30 min to obtain a homogeneous white solution. Subsequently, the resulting solution was transferred to a 50 mL Teflon-lined stainless autoclave and maintained at 150 °C for 24 h in an oven. After cooling down to room temperature, the white precipitates were thoroughly centrifuged and washed three times with H2O and C2H5OH. Finally, the ZnO NRs were collected for further experiments and characterization after drying at 60 °C for 10 h.
2.3 Synthesis of the Au/ZnO and Pt/ZnO NR catalysts
The deposition of Au NPs on the ZnO NRs surface was carried out via a sonochemical method in an ultrasound oscillator (KQ-50E, 50 W) applying HAuCl4·3H2O as the noble metal precursor. Typically, the Au/ZnO catalyst was prepared as follows: the as-prepared ZnO NRs (10 mg) were dispersed in H2O (19 mL). After ultrasonication for 30 min, a desired volume of HAuCl4·3H2O solution with a concentration of 50 mM was added to the uniform ZnO solution. Afterwards, the abovementioned solution was treated under ultrasonic wave irradiation for 2 h after being adequately stirred at room temperature for 5 min. In order to ensure that the temperature of this system was kept at about 30 °C, changing the water every 30 min is indispensable. Two hours later, the as-synthesized purple solid product was collected by centrifugation, washed several times with H2O and C2H5OH, and finally dried at 60 °C in an air atmosphere for 6 h. 50 μL and 100 μL of HAuCl4·3H2O solution were employed to prepared samples with different Au loadings, which were denoted as Au/ZnO-1 and Au/ZnO-2, respectively. The nominal molar ratio of Au to ZnO was 2% and 4% for Au/ZnO-1 and Au/ZnO-2, respectively, which was calculated using the different ratios of HAuCl4·3H2O and ZnO. Similarly, Pt/ZnO-1 and Pt/ZnO-2 were also synthesized via the same procedure except that the noble metal precursor was H2PtCl6·6H2O instead of HAuCl4·3H2O. For comparison, Au surface-modified ZnO catalysts prepared via a chemical reduction method employing NaBH4 as the reductant were denoted as Au/ZnO R1 and R2. The nominal molar ratio of Au to ZnO was 2% and 4% for the Au/ZnO-R1 and Au/ZnO-R2 samples, respectively.
2.4 Characterization
The XRD patterns of the samples were recorded using an X-ray diffractometer (Bruker D8, Germany) in the 2θ range of 20–80° using Cu Kα radiation as a power source. The TEM and HRTEM images of the samples and SAED analysis were performed on a JEOL JEM 2010F microscope to characterize the detailed morphology and crystalline structure of the products. The noble metal loadings of various catalysts were analyzed using energy dispersive X-ray spectrometry (EDX, Oxford Instrument) and X-ray photoelectron spectroscopy (XPS, VG Micro Tech ESCA 3000). The UV-visible diffuse reflectance spectra (DRS) of the obtained photocatalysts were recorded on a Hitachi U-4100 spectrophotometer using BaSO4 as the reference sample. In order to investigate the recombination of photogenerated electrons/holes pairs in the photocatalysts, room-temperature photoluminescence (PL) spectra were obtained on a Hitachi F-4600 PL spectrometer using an excitation wavelength of 325 nm. Photocurrent measurements were carried out using a standard three-electrode cell, consisting of a working electrode (the as-prepared photocatalyst), a platinum wire as counter electrode and Ag/AgCl as the reference electrode. 50 mL of Na2SO4 solution (0.1 M) was used as the electrolyte.
2.5 Photocatalytic performance
The photocatalytic activities of the as-synthesized pure ZnO NRs and Au/ZnO nanostructures were evaluated by the degradation of RhB under UV light irradiation using a 300 W fluorescent Hg lamp with major emission at 365 nm. The photocatalyst (10 mg) was adequately dispersed in an RhB aqueous solution (20 mL, 10 mg L−1). Furthermore, the photocatalytic efficiency under visible light was also investigated employing 2,4-dichlorophenol (20 mL, 10 mg L−1) as the organic pollutant under 300 W Xe lamp irradiation equipped with a cut-off filter (λ ≥ 420 nm). Prior to illumination, the target solution was ultrasonically dispersed for 5 min and then magnetically stirred in the dark for 30 min to reach an adsorption/desorption equilibrium between the photocatalyst and RhB. Afterwards, the abovementioned solution was exposed to light irradiation under continuous stirring. In the process of irradiation, aliquots of 2 mL of the suspension was extracted at regular intervals and centrifuged to remove the photocatalyst. The absorbance spectra of the centrifuged solution were obtained using a Hitachi U-4100 spectrophotometer to calculate the concentration of the pollutant by monitoring its characteristic absorption. The characteristic absorption peaks of RhB and 2,4-dichlorophenol are at 554 nm and 283 nm, respectively. Moreover, the Au/ZnO-2 sample was selected for the photocatalytic stability test under UV light irradiation. The experiment was repeated five times in accordance with the abovementioned photodegradation process.
2.6 CO catalytic oxidation
The catalytic activities of the different catalysts for CO oxidation were evaluated using a fixed-bed flow reactor system under atmospheric pressure. For each test, the catalysts (50 mg) were placed in a quartz tube with an inside diameter of 6 mm. The catalysts were used without any pretreatment before the catalytic oxidation reaction. The reaction gas mixture comprised a volume ratio of 1% CO, 10% O2 and 89% N2, which was allowed to flow through the catalyst sample at a total flow rate of 50 mL min−1. The programmed-temperature rate was controlled at 2 °C min−1 by an electrical furnace and monitored by a thermocouple, which was placed in the middle of the reactor. The compositions of the inlet and outlet gas were analyzed using a gas analyzer. The CO conversion ratio was observed according to the CO consumption and CO2 formation.
3. Results and discussion
3.1 Microstructure and morphology of the Au/ZnO nano heterostructures
The transmission electron microscopy (TEM) images and X-ray diffraction (XRD) patterns of the pure ZnO NRs are shown in Fig. S1 (see ESI).† The XRD pattern demonstrates that all the XRD peaks are consistent with wurtzite ZnO (JCPDS card no. 36-1451) and no characteristic impurity peaks were observed, implying the high purity of the ZnO products. Furthermore, it should be mentioned that the sharp and strong diffraction peaks are ascribed to the as-prepared ZnO with good crystallinity. The SEM and TEM images in Fig. S1b and c† show that the as-prepared ZnO NRs are estimated to be about 25.42 nm in diameter and several hundred nanometers in length. The selected area electron diffraction (SAED) pattern of a single nanorod shown in Fig. S1d† exhibits a series of diffraction spots, signifying its single-crystalline structure.
To synthesize the Au (or Pt) modified ZnO NRs catalysts, a sonochemical method was used in this study without any external reducing agent. According to the reduction reaction of Au3+ ions in the solution with ZnO NRs, the Au NPs or clusters were formed when the initial white suspension turned purple. Similarly, Pt/ZnO-1 and Pt/ZnO-2 heterostructures were also formed, while the color of the ZnO suspension turned light grey after ultrasonic irradiation for 2 h. To confirm the above speculations, TEM, high resolution transmission electron microscopy (HRTEM) and XRD characterizations were performed and are shown below.
The TEM images in Fig. 1a and c show that the Au NPs were randomly distributed on the surface of the ZnO NRs and their size increased with an increasing molar ratio of reactants. The result indicates that the size of hemispherical Au NPs in Au/ZnO-1 was in a range of 4–7 nm. When the nominal molar ratio of HAuCl4 to ZnO was increased to 4%, the diameter of the Au NPs increased to 7–9 nm for Au/ZnO-2 as shown in Fig. 1c. The inset in Fig. 1a shows that dark dots with the size of several nanometers are highly dispersed on the surface of the ZnO NRs as clusters. Similar clusters were also detected for Au/ZnO-2, as shown in Fig. 1c and d. The HRTEM images of the Au/ZnO NR catalysts in Fig. 1b and d exhibit two distinct sets of the lattice fringes. The interplanar spaces were calculated to be 0.26 nm and 0.23 nm, which are consistent with the (002) plane for wurtzite ZnO and the (111) plane for face-centered cubic Au, respectively. These results confirmed the formation of Au NPs on the ZnO NRs. Previously, the hemispherical shape of Au NPs has been observed on a Au/TiO2 system, implying the Au NPs are attached tightly on the ZnO supports.28 This was ascribed to the growth of Au clusters in situ. In our experiment, similar morphological Au NPs were observed. This was ascribed to the sonochemical method that results in the Au NPs being grown tightly on the ZnO NRs. Furthermore, XRD analysis was performed to investigate the composition of the Au/ZnO nanostructures, as shown in Fig. 2e. The result indicates that the two Au/ZnO samples exhibit intense ZnO peaks and a weak Au peak at around 2θ of 38.12°, which further certify the reduction of Au3+ ions under ultrasonic irradiation.
 |
| | Fig. 1 TEM and HRTEM images of the Au/ZnO-1 (a and b) and Au/ZnO-2 (c and d) samples as well as the XRD patterns of the two samples (e). | |
 |
| | Fig. 2 High-resolution XPS spectra of Au4f-Zn3p for Au/ZnO-1 (a) and Au/ZnO-2 (b). | |
Fig. S2 (see ESI†) displays the TEM and HRTEM images of the Pt/ZnO catalysts with different molar ratios of Pt to ZnO NRs. During the ultrasonic reduction, a color change in the solutions was observed when the molar ratio of reactants was less than 2%. However, no visible dark Pt dots are found on the surface of the ZnO NRs, as shown in Fig. S2a† for the Pt/ZnO-1 sample. Only one set of lattice fringes with a spacing value of 0.26 nm was observed from the HRTEM image in Fig. S2b,† which can be assigned to the (002) plane of the ZnO NRs. As reported for supported Au catalysts,2,29 the reason for this was probably due to that the size of the Pt clusters being less than 1 nm. Such small clusters were highly dispersed on the surface of the ZnO NRs and were not detected by TEM. The TEM image in Fig. S2c† shows that Pt clusters with an average size of 2.5 nm were uniformly distributed on the surface of the ZnO NRs when the nominal molar ratio of Pt to ZnO was 4% for the Pt/ZnO-2 sample. The Pt/ZnO-2 catalyst exhibits an interplanar d-spacing of 0.23 nm and 0.26 nm, suggesting the presence of the (111) planes of Pt clusters on the (002) planes of the ZnO surface (Fig. S2d†). The Pt clusters marked by a white arrow can be clearly observed due to their higher electron density than that of the ZnO NRs, as shown in the inset of Fig. S2d.† The abovementioned analysis confirmed the formation of Pt clusters under ultrasonic irradiation in the presence of the ZnO NRs.
To gain further insight into the chemical states of Au in the Au modified ZnO NRs, the samples were studied using XPS. As depicted in Fig. 2, the branches of Au4f7/2 observed for metallic gold were obtained although part of the peaks for Au4f5/2 was overlapped by the signal of Zn3p. The two binding-energy peaks observed at 83.16 and 86.90 eV belong to Au4f7/2 and Au4f5/2 for Au0, respectively, verifying the formation of metallic gold under ultrasonic irradiation. Moreover, the peak located at 85.30 eV is typically associated with Au+, indicating the incomplete reduction of Au3+ in our system. It is observed that the amount of Au0 increased with an increasing molar ratio of the reactants. The actual Au loadings calculated from the XPS spectra were 0.84% and 0.96% for the Au/ZnO-1 and Au/ZnO-2 catalysts, respectively. The values are much lower than those obtained from the EDX spectra shown in Fig. S4,† confirming that the Au3+ ions were partially reduced. Overall, the analyses obtained from TEM, HRTEM, XRD, XPS and EDX not only further confirm the formation of the Au and Pt clusters, but also prove the feasibility of the sonochemical method in the synthesis of Au or Pt surface-modified ZnO catalysts.
3.2 Formation mechanism of the noble metal modified ZnO NRs
As for the in situ growth of the noble metal NPs on ZnO NRs via a sonochemical method, a plausible reduction mechanism for the Au3+ or Pt4+ ions was proposed and shown in Fig. 3. As mentioned previously, researchers have reported that Au NPs were successfully synthesized by the reduction of Au3+ ions in aqueous solutions via a sonochemical reduction method.22–24 In general, an accelerator such as 1-propanol or 2-propanol is essential in the ultrasonic system in order to produce more reducing species. In this study, the reduction of Au3+ or Pt4+ ions can be similarly realized under ultrasonic irradiation without any accelerators in the presence of the ZnO NRs. There are two types of reducing species generated, namely, H atoms and electrons. H atoms are generated from the sonolysis of water. In the sonochemical treatment process, the corresponding reactions occur in three different regions: (1) the inside of the cavitation bubbles, (2) the interface between the cavitation bubbles and the solution, and (3) the aqueous solution at room temperature. The H atoms were created in the collapsing cavitation bubbles because of the high temperature and high pressure environment. The temperature in the interface region was also high enough for the thermal decomposition of H2O. Subsequently, these H atoms can participate in the reduction reaction of Au3+ or Pt4+ by spreading to the surrounding solution.
 |
| | Fig. 3 The proposed reduction mechanism of Au3+ or Pt4+ under ultrasonic irradiation using ZnO NRs as the support material. | |
For the ZnO support materials, electrons in the valence band (VB) can be excited to the conduction band (CB) due to the collapsing of cavitation bubbles producing high energy.25,26 Thus, these free electrons are able to transfer and accumulate on the surface of the ZnO NRs because of the specific piezoelectric effect of the ZnO NRs. Finally, the reduction of Au3+ and Pt4+ was attributed to the synergistic effect of the H atoms and electrons. The major sonochemical reduction of Au3+ proceeds as follows:23
| | |
Au3+ + 3H+ → Au0 + 3H+
| (5) |
The growth of the Au NPs/clusters can be expressed in the following reaction.
In the presence of H2PtCl6, the nucleation and growth reactions of the Pt NPs/clusters are shown below (eqn (9)–(11)).
| | |
Pt4+ + 4H+ → Pt0 + 4H+
| (9) |
where the value of
n in
eqn (8) and
(11) may range from several atoms to thousands, which probably causes the coexistence of metal clusters and NPs.
3.3 Optoelectronic properties of the Au/ZnO NRs
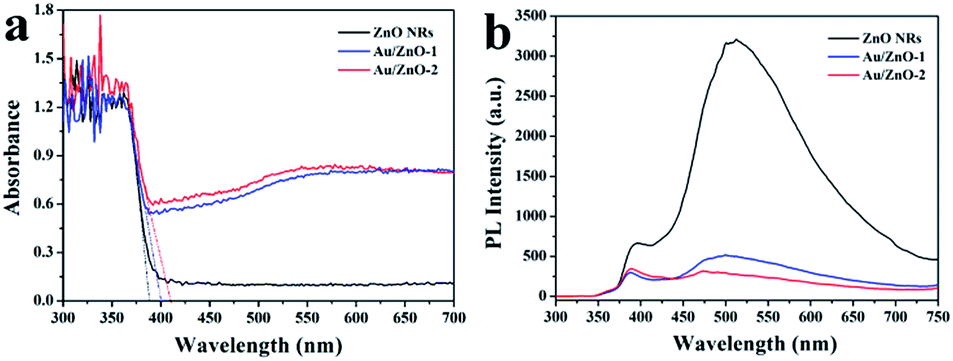
It is well known that the incorporation of Au NPs has a significant impact on the optoelectronic properties of semiconductor nanomaterials due to their unique surface plasmon resonance (SPR) properties.30,31 The optoelectronic properties of the ZnO NRs before and after being decorated with Au NPs/clusters were investigated using UV-visible DRS, PL spectroscopy and photocurrent measurements. Fig. 4a shows that the absorption edges were determined to be about 401 and 410 nm for the Au/ZnO-1 and Au/ZnO-2 samples, respectively, which were shifted towards the visible region when compared with the pure ZnO NRs (388 nm). The red shift of the absorption edge observed for the Au/ZnO NRs was mainly ascribed to the Au NPs. The magnitude of the absorption edge gradually increased upon increasing the Au loading on the ZnO NRs. The absorption edge at the longer wavelength implies that the Au decorated ZnO nanostructures can efficiently utilize light for the photodegradation of pollutants.32 Moreover, the light absorption intensity in the wavelength range from 400 to 700 nm was obviously observed in comparison with the pure ZnO NRs, which was attributed to the band gap transition of the ZnO semiconductor.33 In detail, a Schottky barrier will be formed due to the different work functions of Au and ZnO. In addition, electrons transfer from the Au Fermi level (FM) to the ZnO Fermi level (FMO) because the Fermi level of Au is higher than that of the ZnO support, leading to the formation of a new equilibrium Fermi level. A broad absorption band near 550 nm corresponds to the SPR absorption of Au, suggesting a large size distribution of Au NPs, which is consistent with the TEM results shown in Fig. 1.
 |
| | Fig. 4 (a) UV-visible DRS analysis of pure ZnO NRs, Au/ZnO-1 and Au/ZnO-2 catalysts, and (b) the corresponding PL spectra of the different samples using an excitation wavelength of 325 nm. | |
The PL spectra obtained at room temperature were useful to understand the fate of the photo-generated e−/h+ pairs in the semiconducting materials.9 The PL spectra of the as-prepared ZnO NRs and Au/ZnO NRs were measured using an excitation wavelength of 325 nm, as shown in Fig. 4b. All the catalysts exhibit two prominent bands, a narrow UV emission band at about 390 nm and another broad green band at around 510 nm. The UV emission was assigned to the exciton recombination related near-band edge emission of ZnO,34 whereas the green emission was considered to be associated with an indirect emission, which is related to the surface defects in the ZnO support.35 Obviously, the PL intensity of the Au/ZnO-1 and Au/ZnO-2 samples was drastically quenched when compared with the pure ZnO NRs, illustrating that direct and defect-related charge carrier recombination pathways were blocked by Au NPs/clusters. In addition, the effect of the Au loading on the PL intensity was negligible due to the highly dispersed Au clusters on the surface of the ZnO NRs. These results indicate that the Au NPs/clusters enable the capture of electrons from the conduction band of ZnO that effectively inhibits the recombination process of the photo-generated e−/h+ pairs, leading to higher photocatalytic activity.
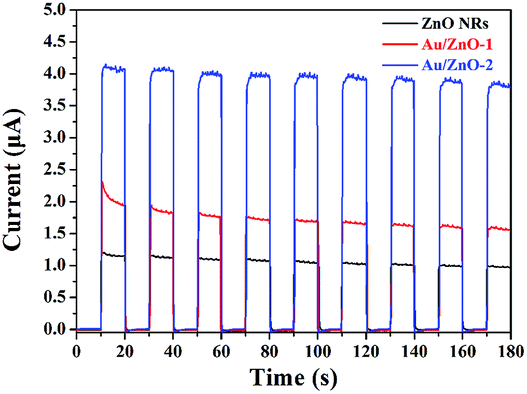
It is widely accepted that the separation efficiency of the e−/h+ pairs plays a crucial role in improving the photocatalytic activity.36 The generation and transfer of photo-induced charge carriers in the photocatalytic process can be monitored by photocurrent measurements. In general, a higher value of the photocurrent shows a higher efficiency of e−/h+ pairs separation.37 Fig. 5 shows the transient photocurrent response of the ZnO NRs, Au/ZnO-1 and Au/ZnO-2 electrodes. The result indicates that the photocurrent trend follows the order Au/ZnO-2 > Au/ZnO-1 > ZnO NRs. The value of the photocurrent was significantly enhanced upon introducing the Au NPs/clusters onto the surface of the ZnO NRs, implying that the photogenerated e−/h+pairs in the Au/ZnO NRs have a longer lifetime than that of the pure ZnO NRs. The Au/ZnO-2 sample exhibits the highest photocurrent response, which is about 3.4 times higher than that observed for the pure ZnO NRs, suggesting the maximum separation efficiency of the photo-generated carriers.
 |
| | Fig. 5 The UV photocurrents of ZnO NRs, Au/ZnO-1 and Au/ZnO-2 electrodes towards irradiation with 365 nm light at room temperature (0.1 M Na2SO4). | |
3.4 Photocatalytic properties and mechanism of the Au/ZnO catalysts
It is obvious that the Au/ZnO NRs have promising photocatalysis applications according to the above discussion. Photocatalytic tests were carried out by employing an aqueous solution of RhB dye as a model contaminant. Prior to light exposure, all the samples were kept in the dark for 30 min to reach an adsorption/desorption equilibrium. The percentage of RhB adsorption was measured to be 10.88%, 3.85% and 11.03% for the ZnO NRs, Au/ZnO-1 and Au/ZnO-2, respectively. Fig. S5 (see ESI†) shows the temporal absorption results for RhB using Au/ZnO-2 as a catalyst. With increasing time, the intensity of the characteristic absorption peak at 550 nm gradually decreased, indicating that the RhB was photodegraded. It can be noted that RhB was almost completely degraded after only 12 min using the Au/ZnO-2 sample, which was significantly faster than those previously reported.27,38,39
The degradation efficiency is defined as C/C0, where C0 and C represent the concentration of RhB after 30 min dark treatment and the instantaneous concentration of RhB during the photodegradation process, respectively. To quantitatively analyse the photodegradation behavior, the degradation efficiency (C/C0) of the different catalysts was investigated and shown in Fig. 6a. No obvious degradation with the blank test (no catalysts) was observed, showing that photoinduced self-sensitized photolysis can be ignored in contrast to the photocatalysis in the presence of catalysts. The photocatalytic performance of pure ZnO NRs was unpropitious, affording only 84% degradation of RhB under UV illumination after 60 min. However, the introduction of Au NPs/clusters accelerates the photodegradation of RhB, certifying that integrating ZnO NRs with noble metals is promising to improve the degradation efficiency. When compared with pure ZnO NRs, the Au/ZnO-1 sample has a degradation efficiency of about 99.9% after irradiation for 24 min, whereas the efficiency of the Au/ZnO-2 sample reaches nearly 100% only after 12 min. In contrast, a degradation of nearly 100% occurred within 50 min for Au/ZnO-R1 and R2. Obviously, the degradation efficiency of the Au/ZnO nanostructures prepared by the sonochemical reduction method was much higher than that observed for the Au/ZnO-R composite obtained by a chemical reduction method. The kinetics of photodegradation for the different catalysts was explored to further understand the improved photocatalytic efficiency in the abovementioned photocatalysis.
 |
| | Fig. 6 (a) A comparison of the photocatalytic efficiency in the presence of ZnO NRs, Au/ZnO-1, Au/ZnO-2, Au/ZnO-R1, Au/ZnO-R2 and no catalyst under UV light irradiation. (b) A plot of ln(C0/C) as a function of UV irradiation time for the different degradation systems. (c) The degradation dynamics of RhB using Au/ZnO-2 as the photocatalyst for 5 cycles of recycled use. (d) The photocatalytic degradation profile toward 2,4-dichlorophenol under visible-light irradiation. | |
As shown in Fig. 6b, it was clearly observed that the photodegradation data of RhB fit well to pseudo-first-order reaction model kinetics:26 ln(C0/C) = kt, where k is the photodegradation rate constant and t denotes the UV light irradiation time. The value of k was equal to the corresponding slope of the pseudo-first-order linear simulation curve. The degradation rate constant k observed for the different photocatalysts followed the order 0.332 min−1 (Au/ZnO-2) > 0.147 min−1 (Au/ZnO-1) > 0.083 min−1 (Au/ZnO-R2) > 0.076 min−1 (Au/ZnO-R1) > 0.030 min−1 (ZnO NRs). For Au/ZnO-1, the k value was about 5 times higher than that found for pure ZnO NRs. In particular, the Au/ZnO-2 sample shows the highest value for the rate constant, which was almost 11 times greater than that found for the ZnO NRs. Herein, it can be directly observed that the photodegradation efficiency of Au/ZnO was much higher than that of the pure ZnO NRs due to the faster separation efficiency of the photoinduced electrons and holes in the Au/ZnO catalysts.
Maintaining the recyclability and high photocatalytic activity of a photocatalyst are two extremely important criteria in practical applications. The reusability of the Au/ZnO-2 catalyst was evaluated for the degradation of RhB dye in five repeated cycles under the same conditions. The photocatalyst was centrifuged and washed three times with H2O before use in each cycle. As displayed in Fig. 6c, the photocatalytic efficiency of Au/ZnO-2 does not display a significant decrease (only 11% reduction) after reusing five times, suggesting that the Au/ZnO-2 photocatalyst exhibits sufficient stability and high activity during the photocatalytic reaction. The above results demonstrate that the Au/ZnO nanostructures synthesized under ultrasonic irradiation have practical value due to their high photodegradation activity and excellent recyclability.
It is satisfactory that the as-obtained Au/ZnO catalysts exhibit photocatalytic activity under visible light towards 2,4-dichlorophenol. As shown in Fig. 6d, an ∼75% degradation of 2,4-dichlorophenol was achieved after 4 h using Au/ZnO-1. Moreover, the degradation can reach about 68% when employing Au/ZnO-2 as the catalyst. The abovementioned results indicate that the introduction of Au NPs was beneficial to broadening the application area of pure ZnO.
According to the abovementioned results, a schematic of the photocatalytic mechanism employing Au/ZnO as a photocatalyst is illustrated in Fig. 7. According to previous studies, the work functions of ZnO and Au were reported to be 5.3 and 5.1 eV, respectively, suggesting a lower FMO for ZnO in contrast to the FM observed for Au (Fig. 7a). In the case of the pure ZnO NRs, only under UV light irradiation, the electrons (e−) in the VB of ZnO are excited and jump to the CB, leaving behind equivalent holes (h+) in the VB. These photoexcited electrons can reduce dissolved oxygen to generate superoxide radical anions (O2˙−), whereas the photoexcited holes in the VB can oxidize OH− and H2O to produce hydroxyl radicals (˙OH). These highly active radicals are both capable of destroying dye molecules, resulting in the generation of CO2 and H2O.40 When Au was introduced onto the surface of the ZnO NRs, a metal–semiconductor Schottky barrier was formed due to the lower Femi level of ZnO when compared to metallic Au. As shown in Fig. 7b, the photogenerated electrons in the CB of the ZnO NRs are captured by Au due to the higher Fermi level of Au under UV light irradiation. Therefore, the Au NPs/clusters usually act as an electron trap in Au modified ZnO nanostructures.41 However, the Schottky barrier prevents the transfer of electrons from Au to the ZnO support. This charge separation process significantly increases the lifetime of the photogenerated carriers in the Au/ZnO catalysts, inducing a faster photodegradation efficiency when compared to the bare ZnO semiconductor. Fig. 7c shows the degradation mechanism of the Au decorated ZnO NRs under the visible irradiation. Au NPs can absorb the resonant photons to generate hot electrons because of SPR excitation. The electrons on the surface of the Au NPs were quickly injected to the CB of ZnO. The accumulated electrons can react with the dissolved oxygen to produce reactive radical species, which are responsible for the decomposition of 2,4-dichlorophenol under visible light irradiation.
 |
| | Fig. 7 A schematic of the band configurations of ZnO and Au (a). A schematic of the enhanced photocatalytic activity of the Au modified ZnO NRs under UV (b) and visible light (c) irradiation. | |
3.5 CO oxidation performance of the Au/ZnO NR catalysts
We further investigated the effects of noble metals (Au, Pt) decoration on the CO oxidation performance of the ZnO support. For comparison, the pure ZnO NRs were also characterized during the CO catalytic oxidation process. As shown in Fig. 8, the conversion ratio gradually increases with the reaction temperature, indicating that CO oxidation occurs continually. Moreover, the CO oxidation data obtained for the pure ZnO NRs, as well as Au and Pt doped ZnO NRs, are summarized in Table 1. Herein, the temperature of the CO conversion of 50% and complete oxidation are denoted as T50 and T100, respectively. The ZnO NRs showed poor catalytic activity towards CO oxidation, which indicates that pure ZnO was inactive as the temperature was below 250 °C. According to previous reports, ZnO is only a good support for noble metals deposition due to its acidic (Zn2+) and basic (O2−) surface sites.42 When the Au or Pt NPs/clusters were introduced to the surface of the ZnO NRs, the CO catalytic activity order was found to be in the following order: Pt/ZnO-2 > Pt/ZnO-1 > Au/ZnO-2 > Au/ZnO-1 > ZnO NRs. Obviously, the CO oxidation activity of the Pt/ZnO samples was much higher than that of the Au/ZnO samples, which was attributed to the Pt clusters with small size being well dispersed on the surface of the ZnO NRs. Furthermore, it can be observed that the CO catalytic activity of Au/ZnO or Pt/ZnO was gradually improved with an increasing amount of Au or Pt due to the increasing number of active sites available for the CO catalytic oxidation on Au/ZnO-2 and Pt/ZnO-2. In particular, the values of T50 and T100 for the Au/ZnO-2 sample decreased to 222 °C and 237 °C, respectively. For Pt/ZnO-2, the value of T100 was dramatically decreased to 176 °C from 279 °C for the pure ZnO NRs, indicating that Pt/ZnO-2 has great potential in practical applications. To explore the optimal loading amount of Au and Pt, the Au/ZnO and Pt/ZnO composites were also prepared by adjusting the volume of precursor solution to 200 μL. The obtained products were denoted as Au/ZnO-3 and Pt/ZnO-3. The CO oxidation results obtained for Au/ZnO-3 as well as Pt/ZnO-3 are shown in Fig. S6 and summarized in Table S1.† The general trend was clear; the catalytic activity was gradually enhanced upon increasing the amount of Au or Pt. The significantly enhanced CO catalytic activity was mainly ascribed to the strong interfacial interactions between the noble metals (Au and Pt) and the ZnO support, which has been demonstrated in the literature.1,9
 |
| | Fig. 8 CO conversion as a function of reaction temperature for CO oxidation on the pure ZnO NRs, Au/ZnO-1, Au/ZnO-2, Pt/ZnO-1 and Pt/ZnO-2 catalysts. | |
Table 1 Conversion temperature of CO using pure ZnO NRs, and Au/ZnO, Pt/ZnO NRs catalysts
| Sample |
ZnO |
Au/ZnO-1 |
Au/ZnO-2 |
Pt/ZnO-1 |
Pt/ZnO-2 |
| T50 [°C] |
277 |
267 |
222 |
179 |
169 |
| T100 [°C] |
279 |
277 |
237 |
185 |
176 |
4. Conclusions
Au/ZnO NR catalysts were synthesized via an ultrasound irradiation method using a solution containing ZnO NRs. During the ultrasonic irradiation, sonolysis of H2O can lead to the formation of H atoms in the cavitation bubbles and the interface between these bubbles and the solution. Upon the collapse of the cavitation bubbles, the released energy is high enough to excite electrons from the VB to CB of ZnO. The H atoms and free electrons are capable of reducing Au3+ to Au NPs/clusters on the ZnO surface. It was found that the Au/ZnO NRs were much more active when compared with pure ZnO NRs towards the photocatalytic degradation of RhB. The highest photocatalytic efficiency reached up to 100% after UV light illumination for 12 min using Au/ZnO-2. Supported by the photocurrent and PL spectra results, the reason for this was mainly attributed to the Au NPs capturing the electrons, which delays the recombination process of the photogenerated charge carriers. Furthermore, the Au/ZnO-2 sample possessed good stability in the photocatalytic process. The sonochemical method employed in this work is also applicable for the preparation of Pt/ZnO composites. A systematic study of CO oxidation demonstrated that the ZnO-supported Au and Pt catalysts show significantly higher catalytic activity. This improved activity was interpreted as a result of the strong interfacial interactions between the noble metal NPs (Au and Pt) and the ZnO support. This study provides a novel sonochemical method to synthesize Au/ZnO and Pt/ZnO NR catalysts with significantly improved catalytic activity for dye degradation and CO oxidation.
Acknowledgements
This study was supported by the Program for Taishan Scholars and the National Natural Science Foundation of China (Grant No. 51572109, 51501071, 51302106, 51402123 and 51402124).
Notes and references
- X. Liu, M. H. Liu, Y. C. Luo, C. Y. Mou, S. D. Lin, H. Cheng, J. M. Chen, J. F. Lee and T. S. Lin, J. Am. Chem. Soc., 2012, 134, 10251 CrossRef CAS PubMed.
- K. Zhao, H. Tang, B. Qiao, L. Li and J. Wang, ACS Catal., 2015, 5, 3528 CrossRef CAS.
- W. Xing, G. Chen, C. Li, J. Sun, Z. Han, Y. Zhou, Y. Hu and Q. Meng, ChemCatChem, 2016, 8, 1 CrossRef.
- L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644 CrossRef CAS PubMed.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752 CrossRef CAS PubMed.
- X. Guo, C. Mao, J. Zhang, J. Huang, W. Wang, Y. Deng, Y. Wang, Y. Cao, W. Huang and S. Yu, Small, 2012, 8, 1515 CrossRef CAS PubMed.
- G. C. Bond and D. T. Thompson, Gold Bull., 2000, 33, 41 CrossRef CAS.
- S. A. C. Carabineiro, B. F. Machado, R. R. Bacsa, P. Serp, G. Dražić, J. L. Faria and J. L. Figueiredo, J. Catal., 2010, 273, 191 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170 CrossRef CAS.
- S. Chatterjee, K. Bhattacharyya, P. Ayyub and A. K. Tyagi, J. Phys. Chem. C, 2010, 114, 9424 CAS.
- J. Wang, J. Liu, H. Xu, S. Ji, J. Wang, Y. Zhou, P. Hodgsonc and Y. Li, J. Mater. Chem. A, 2013, 1, 1117 CAS.
- Z. B. Yu, Y. P. Xie, G. Liu, G. Q. Lu, X. L. Ma and H. M. Cheng, J. Mater. Chem. A, 2013, 1, 2773 CAS.
- L. Zhang, L. Du, X. Yu, S. Tan, X. Cai, P. Yang, Y. Gu and W. Mai, ACS Appl. Mater. Interfaces, 2014, 6, 3623 CAS.
- S. Zhang, H. S. Chen, K. Matras-Postolek and P. Yang, Phys. Chem. Chem. Phys., 2015, 17, 30300 RSC.
- Y. K. Mishra, G. Modi, V. Cretu, V. Postica, O. Lupan, T. Reimer, I. Paulowicz, V. Hrkac, W. Benecke, L. Kienle and R. Adelung, ACS Appl. Mater. Interfaces, 2015, 7, 14303 CAS.
- F. Lu, W. Cai and Y. Zhang, Adv. Funct. Mater., 2008, 18, 1047 CrossRef CAS.
- H. Zhang, G. Chen and D. W. Bahnemann, J. Mater. Chem., 2009, 19, 5089 RSC.
- N. T. Khoa, S. W. Kim, D. H. Yoo, S. Cho, E. J. Kim and S. H. Hahn, ACS Appl. Mater. Interfaces, 2015, 7, 3524 CAS.
- P. Li, Z. Wei, T. Wu, Q. Peng and Y. Li, J. Am. Chem. Soc., 2011, 133, 5660 CrossRef CAS PubMed.
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421 CrossRef CAS.
- S. Zhang, L. Wang, Y. Zeng, Y. Xu, Y. Tang, S. Luo, Y. Liu and C. Liu, ChemCatChem, 2016, 8, 2557 CrossRef CAS.
- K. Okitsu, A. Yue, S. Tanabe, H. Matsumoto and Y. Yobiko, Langmuir, 2001, 17, 7717 CrossRef CAS.
- R. A. Caruso, M. Ashokkumar and F. Grieser, Langmuir, 2002, 18, 7831 CrossRef CAS.
- K. Okitsu, M. Ashokkumar and F. Grieser, J. Phys. Chem. B, 2005, 109, 20673 CrossRef CAS PubMed.
- Y. Liu, M. Zhong, G. Shan, Y. Li, B. Huang and G. Yang, J. Phys. Chem. B, 2008, 112, 6484 CrossRef CAS PubMed.
- S. Khanchandani, S. Kumar and A. K. Ganguli, ACS Sustainable Chem. Eng., 2016, 4, 1487 CrossRef CAS.
- J. Kim and K. Yong, J. Nanopart. Res., 2012, 14, 1 Search PubMed.
- T. Akita, P. Lu, S. Ichikawa, K. Tanaka and M. Haruta, Surf. Interface Anal., 2001, 31, 73 CrossRef CAS.
- K. Zhao, B. Qiao, J. Wang, Y. Zhang and T. Zhang, Chem. Commun., 2011, 47, 1779 RSC.
- Z. Bian, T. Tachikawa, W. Kim, W. Choi and T. Majima, J. Phys. Chem. C, 2012, 116, 25444 CAS.
- W. He, H. K. Kim, W. G. Wamer, D. Melka, J. H. Callahan and J. J. Yin, J. Am. Chem. Soc., 2014, 136, 750 CrossRef CAS PubMed.
- C. Shifu, C. Lei, G. Shen and C. Gengyu, Powder Technol., 2005, 160, 198 CrossRef.
- M. Wu, W. J. Chen, Y. H. Shen, F. Z. Huang, C. H. Li and S. K. Li, ACS Appl. Mater. Interfaces, 2014, 6, 15052 CAS.
- Y. Hu, Z. Jiang, C. Xu, T. Mei, J. Guo and T. White, J. Phys. Chem. C, 2007, 111, 9757 CAS.
- J. Wang and L. Gao, Solid State Commun., 2004, 132, 269 CrossRef CAS.
- T. L. Thompson and J. T. Yates, Chem. Rev., 2006, 106, 4428 CrossRef CAS PubMed.
- H. W. Huang, Y. He, Z. S. Lin, L. Kang and Y. H. Zhang, J. Phys. Chem. C, 2013, 117, 22986 CAS.
- W. L. Ong, S. Natarajan, B. Kloostra and G. W. Ho, Nanoscale, 2013, 5, 5568 RSC.
- C. Yu, K. Yang, Y. Xie, Q. Fan, J. C. Yu, Q. Shu and C. Wang, Nanoscale, 2013, 5, 2142 RSC.
- Y. Zheng, C. Chen, Y. Zhan, X. Lin, Q. Zheng, K. Wei and J. Zhu, J. Phys. Chem. C, 2008, 112, 10773 CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Phys. Chem. B, 2003, 107, 7479 CrossRef CAS.
- E. Castillejos, R. Bacsa, A. Guerrero-Ruiz, I. Rodríguez-Ramos, L. Datas and P. Serp, Nanoscale, 2011, 3, 929 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra23471b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.