
Zeylenone promotes apoptosis in chronic myelogenous leukemia-derived K562 cells by a mechanism involving Jak2 and Src kinase†
Xiaowei Huo ,
Yonghong Liao,
Yu Tian,
Li Gao and
Li Cao*
,
Yonghong Liao,
Yu Tian,
Li Gao and
Li Cao*
Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100193, China. E-mail: lcao@implad.ac.cn; Tel: +86-10-57833222
First published on 30th November 2016
Abstract
Chronic myelogenous leukemia (CML) is a hematopoietic malignancy caused by the constitutive activation of BCR–ABL tyrosine kinase. Imatinib, an inhibitor of BCR–ABL, is very effective in controlling CML, however, the emergence of resistance remains a concern. Therefore, alternative strategies with unique targets are strongly desirable. Zeylenone (Zey), isolated from the leaves of Uvaria grandiflora Roxb. of the family Annonaceae, exhibits significant anticancer activity in various types of cancer cells. However, the anticancer effect of Zey on CML and the mechanism of action remains unclear. Thus, the present study was designed to investigate the anticancer effect of Zey on K562 cells both in vitro and in vivo, followed by exploring the underlying mechanisms. Initially, the effects of Zey on cell viability, proliferation, and apoptosis were measured in K562 cells after they were treated with Zey for the indicated time, and then the involved signaling pathways were investigated. Furthermore, the in vivo anti-tumor activity of Zey was assessed with nude xenografts. We identified that Zey dose-dependently decreased cell viability and proliferation, and induced cell apoptosis in K562 cells. Additional experiments revealed that Zey inhibited phosphorylation of Jak2 and Src and downregulated their downstream proteins, including p-Stat3, p-PI3K, p-AKT, p-mTOR, and p-ERK. Zey also suppressed tumor growth with low toxicity in a mouse xenograft model of K562 cells. Taken together, our data demonstrated that Zey substantially suppressed K562 cells both in vitro and in vivo through Jak2 and Src pathways. These findings suggest the potential of zeylenone as an effective anticancer agent in CML treatment.
Background
Chronic myeloid leukemia (CML) is a hematopoietic stem cell malignancy initiated by the oncogenic fusion protein BCR–ABL, a deregulated, aberrantly activated tyrosine kinase driven by chromosomal reciprocal translocation between chromosome 22 and chromosome 9 (t(9; 22) (q34; q11)), also known as the Philadelphia chromosome.1–4 The BCR–ABL kinase recruits and constitutively activates multiple pathways, including Ras, signal transducer and activator of transcription (STATs), extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK), phosphatidylinositol-3 kinase (PI3K) and nuclear factor (NF)-κB, that transduce oncogenic signals, thus leading to uncontrolled proliferation, inhibition of apoptosis, and block of myeloid differentiation in myeloid lineage cells.5,6 Although allogeneic stem cell transplantation has been considered the only curative therapy for CML, it is only available to a minority of individuals because most of them lack suitable donors or are not eligible for transplant due to advanced age.7,8The clinically use of imatinib myselate (aka STI571, Gleevec), a small molecule inhibitor of the BCR–ABL kinase, has met with much success in the control of newly diagnosed CML patients, leading to sustained cytogenetic and molecular remissions in majority of patients with chronic-phase CML.9–11 However, most patients with progression beyond the chronic phase are prone to relapse after an initial response due to the occurrence of imatinib resistance mainly caused by ABL kinase domain mutations that impair drug binding either directly or through allosteric mechanisms, and amplification or over expression of BCR–ABL,12 thus sustained remission of BCR–ABL by imatinib treatment becomes a challenge. Imatinib failure then results in the development and rapid approval of nilotinib and dasatinib, the second-generation inhibitors targeting most imatinib–resistant BCR–ABL variants,13 however, resistance and unsatisfactory responses in advanced disease stages of all three BCR–ABL inhibitors remain major problems for their clinical use.14,15 Thereby, discovery of new agents with novel therapeutic targets besides BCR–ABL is imperative for the treatment of CML.
Naturally occurring bioactive phytochemicals with low-toxic nature have emerged as promising options for the development of effective alternatives for conventional cytotoxic treatments.16 Zeylenone, a naturally occurring cyclohexene oxide, isolated from ethanol extract of the leaves of Uvaria grandiflora Roxb. of the family Annonaceae, exhibited significant anticancer activity in several human cancer cells, including breast, prostate and hepatocellular carcinoma, with no apparent signs of toxicity on normal cell lines.17 Previous studies by our group demonstrated that Zey inhibited proliferation of acute lymphoblastic leukemia Reh and RS4; 11 cells in a dose-dependent manner,18 with low toxicity against normal peripheral blood mononuclear cell (PBMC) (ESI† data), indicating its potential as an anti-leukemia agent. Nevertheless, there is only limited information regarding the effects of this compound on CML, and the mechanisms responsible for its anticancer effect are less well understood.
The purpose of the present study is to investigated the anticancer effects of Zey on CML cell line K562 both in vitro and in vivo, and further explore the underlying mechanisms. Our data demonstrated that Zey induced substantial apoptosis of K562 cells, which was associated with the mitochondrial apoptotic pathway. Further studies revealed that these anticancer effects of Zey possibly involve a mechanism of inhibiting phosphorylation of Jak2 and Src kinase, and thus the corresponding downstream signaling pathways including Stat3 and PI3K/AKT/mTOR and ERK1/2. In vivo assays with K562 xenografts model confirmed anticancer effects of Zey and verified the dual inhibition of p-Jak2 and p-Src by Zey treatment. Thus, we conclude that Zey has potential for development of new therapeutic agents for human CML treatment.
Methods
Reagents
Preparations of zeylenone and mPEG-PLGA loaded zeylenone micelles were described previously.19 Zeylenone used for in vitro study was stored as 100 mM solutions in DMSO at −20 °C and further diluted to desired working concentration before each use (DMSO concentration < 1‰). mPEG-PLGA loaded zeylenone micelles used for in vivo study was stored in a dry container at room temperature. RPMI-1640 and fetal bovine serum (FBS) were purchased from Gibco (CA, USA), Antibodies against PI 3-kinase p110 (A-8), p-PI 3-kinase p85α (Tyr 467), Lamin B (B-10), XIAP (H-202), survivin (FL-142), AIF (B-9), Bcl-2, Bcl-XL, p-ERK Antibody (E-4), PCNA (PC10), p-JNK (G-7) and PARP-1 (H-250) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against phospho-Jak2 (Tyr1007) (D15E2), phospho-Stat3 (Tyr705) (M9C6), phospho-Src (Tyr527), phospho-Akt (Ser473) (D9E), Akt (pan) (40D4), phospho-mTOR (Ser2448), PI3 kinase p110α (C73F8), PI3 kinase p110β (C33D4), PI3 kinase p110γ (D55D5), phospho-PTEN (Ser380/Thr382/383) (44A7), and β-actin were purchased from Cell Signaling Technology (Danvers, MA). Methyl thiazolyl tetrazolium (MTT), 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide (JC-1), acridine orange (AO), and ethidium bromide (EB) were purchased from Sigma Aldrich Chemical (St. Louis, MO). Imatinib were purchased from Selleck Chem (Houston, TX). Annexin V-FITC kit was obtained from BD Biosciences (San Jose, CA).Cell lines and cell culture
Human K562 cells were obtained from Chinese Academy of Medical Sciences Basic Medicine Cell Center (Beijing, China). Cells were cultured in RPMI-1640 media containing 10% fetal bovine serum (FBS), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin at 37 °C in an atmosphere of 5% CO2.Cell viability
K562 cells cultured in 96 well plates at a density of 6 × 103 cells per well were treated with different concentrations of Zey for 24 h, and cell viability was monitored by MTT assay, as previously described.20 After incubation for 24 h, 10 μL MTT reagent (5 mg mL−1) was added to the media and allowed to incubate for another 4 h at 37 °C in a humidified incubator at 5% CO2. The medium was then removed and MTT crystals were dissolved in 100 μL DMSO. Cell viability was quantified by reading the plates at an absorbance of 540 nm using a microplate reader. The 540 nm absorbance of vehicle-treated wells was used to define 100% proliferation. The concentration of drug that resulted in 50% growth inhibition or loss of viability when compared to vehicle treated control cells was calculated and reported as the IC50 value. Each condition was performed in triplicates of six wells.Soft agar assay
Six-well plates were covered with 1 mL of 0.6% agar in medium supplemented with 20% fetal bovine serum. K562 cells (1 × 104) were prepared in 0.3% agar containing Zey at the indicated concentrations (0, 0.4, 0.8, and 1.6 μM), and seeded in triplicate. The cells were allowed to incubate at 37 °C for 2 weeks, and then colonies were counted as previously described.21 Cell aggregates containing 40 or more cells were considered colonies. Each experiment was performed in triplicate.Observation of apoptotic cell morphology
K562 cells after treatment with Zey at different concentrations (0, 0.4, 0.8, and 1.6 μM) were observed by inverted microscopy (Olympus, Tokyo, Japan), images were then captured.AO/EB staining
K562 cells were seeded at 1 × 105 cells per mL in 6-well plates and treated with different concentrations of Zey (0, 0.4, 0.8, and 1.6 μM) for 24 h. After washing with phosphate-buffered saline (PBS), cells were stained with dye mixture containing 100 mg L−1 AO and 100 mg L−1 EB solution of each dye and should be kept away from light exposure. Microscope images were captured using EVOS FL imaging system.Flow cytometric analysis of apoptosis
K562 cells cultured in 6 well plates were treated with vehicle alone or Zey as indicated (0.4, 0.8, and 1.6 μM) for 24 h. Cells were then collected, washed with PBS, and stained with annexin-V-FITC for 15 min in the dark before propidium iodide was added. The stained cells (104 cells) were then analyzed immediately using a FACS Calibur cytometer (Becton Dickinson, CA, USA) and results were expressed as percentage of living (AnnV-, PI-), early apoptotic (AnnV+, PI-), and late apoptotic/dead cells (AnnV+, PI+). Apoptotic rates were reported as the percentage of apoptotic cells among total cells. All measurements were made in triplicate and averaged.JC-1 for mitochondrial transmembrane potential study
K562 cells were incubated in the presence of various concentrations of Zey (0, 0.4, 0.8, and 1.6 μM) for 24 h, and were then incubated with 10 μg mL−1 JC-1 for 30 min at 37 °C. The cells were collected, washed with PBS, and analyzed on a flow cytometer. Highly negative membrane potential in mitochondria produces JC-1 red fluorescence. Loss of mitochondrial transmembrane potential results in green fluorescence and loss of the red fluorescence.RNA isolation and real-time quantitative polymerase chain reaction (RT-qPCR) analysis
Total RNA from cells was extracted using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. The cDNA was synthesized using the Superscript First-Strand Synthesis kit (Aidlad, Beijing, China) and used as template for real-time PCR with a sequence specific primer and SYBR Green PCR Master Mix kit (Cowin Biotech Co., Ltd., Beijing, China) at the following thermal conditions: 95 °C for 10 min and 40 cycles at 95 °C for 5 s and 60 °C for 32 s. Relative mRNA expression was calculated by the 2−ΔCt method [ΔCt = Ct (protein) − Ct (beta-actin)] with beta-actin for normalization. All samples were performed in triplicate for three times. The primers for RT-qPCR are as follows: survivin forward, 5′-AGC CCT TTC TCA AGG ACC AC-3′; reverse, 5′-GCA CTT TCT TCG CAG TTT CC-3′. XIAP forward, 5′-CCC AAA TTG CAG ATT TAT CAA CG-3′; reverse, 5′-TGC ATG TGT CTC AGA TGG CC-3′. Bcl-xL forward, 5′-TAT TGG TGA GTC GGA TCG CAG CTT-3′; reverse, 5′-TGG TCA GTG TCT GGT CAT TTC CGA-3′. Bcl-2 forward, 5′-GGA TTG TGG CCT TCT TTG AG-3′; reverse, 5′-CCA AAC TGA GCA GAG TCT TC-3′. β-Actin forward, 5′-CAC GAT GGA GGG GCC GGA CTC ATC-3′; reverse, 5′-TAA AGA CCT CTA TGC CAA CAC AGT-3′.Western blot analysis
Zey treated and untreated cells (1.5 × 107) were lysed with ice-cold RIPA buffer containing protease and phosphatase inhibitor cocktail purchased from Cowin Biotech (Beijing, China). Protein concentration was determined using a BCA protein estimation kit (Cowin Biotech Co., Ltd., Beijing, China). Equal amount of sample lysate were separated by SDS-PAGE and transferred to PVDF membrane. The membrane was blocked with 5% nonfat milk in TBST for 2 h followed by probed with primary antibody overnight at 4 °C. After washing with TBST, the membrane was subsequently incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies, washed with TBST, and then detected by enhanced chemiluminescence method using a commercial ECL kit (Cowin Biotech Co., Ltd.), according to manufacturer's instructions. The levels of protein expression were quantified by image J software (National Institutes of Health, Bethesda, MD, USA) and normalized to the relative internal standards. All of the experiments were performed in triplicate.Immunofluorescence
K562 cells were cultured in 6-well plates and treated with or without Zey for 24 h. The cells were collected and fixed in 4% paraformaldehyde (PFA) for 20 min, permeabilized with 0.5% Triton X-100 for 15 min at room temperature. After washing twice with PBS, the cells were blocked with 5% Bovine Serum Albumin (BSA) for 1 h at room temperature, and then incubated at 4 °C overnight with primary antibodies including PCNA, p-Jak2 and p-Src at a dilution of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100. The cells were washed twice with PBS and incubated with PE-conjugated secondary antibody for 1 h at room temperature. They were also stained with Hoechst 33258 to visualize the nuclei. The cells were then washed twice with PBS, followed by detection using EVOS FL imaging system.
:100. The cells were washed twice with PBS and incubated with PE-conjugated secondary antibody for 1 h at room temperature. They were also stained with Hoechst 33258 to visualize the nuclei. The cells were then washed twice with PBS, followed by detection using EVOS FL imaging system.
Tumor xenograft study
Female nude balb/c mice purchased from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) were maintained and treated according to protocols (no. SLXD-16-01-15) approved by the Institutional Animal Care and Use Committee at the Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences. A total of 1 × 107 of K562 cells were inoculated subcutaneously on the right flanks of mice at age about 5 weeks (5 mice per group). The tumor xenografts were monitored every 2 day with an electronic caliper measurements and tumor volume (V) was calculated using the formula V = (L × W2)/2, where L is the largest diameter and W is the diameter perpendicular to width. When the tumors reached 50–100 mm3, the mice were randomized and treated with vehicle (saline), mPEG-PLGA loaded zeylenone micelles (30 mg per kg per day, i.p.), and imatinib (50 mg per kg per day, i.p.) every day for a total of 14 days. Meanwhile, mouse body weight was measured every 2 days. Mice were then euthanized and tumor xenografts, kidney, liver, and spleen were removed, weighed, stored, and fixed.Immunohistochemical (IHC) analysis
IHC analyses to detect phosphorylation levels of Jak2, Src and Stat3 in the xenograft tumors were performed as described previously.22 After fixed with 4% paraformaldehyde, the tissue were dehydrated in a graded series of alcohol, and then embedded in paraffin. The tissue sections (3 μm) were deparaffinized, dehydrated, and treated with 3% hydrogen peroxide for 10 min to quench the endogenous peroxidase. After consecutive washing with PBS, slides were incubated with p-Stat3, p-Jak2, and p-Src antibodies at a dilution of 1:50 at 4 °C overnight, followed by incubation with biotinylated secondary antibodies (1:100) for 1 hour at room temperature. Signals were detected using Diaminobenzidine Substrate kit (Vector Laboratories). The Image-ProPlus 4.5 Software (Media Cybernetics) was used to analyze the expression of proteins.
Histopathological analysis
Formalin-preserved kidney, liver, and spleen were dehydrated and embedded in paraffin by standard techniques. Paraffin embedded samples were then sectioned at 3 μm, stained with hematoxylin and eosin (H&E) and examined by a pathologist blinded to the experimental groups.Statistical analysis
All experiments were performed in triplicate, and results are expressed as mean ± SD where applicable. GraphPad Prism 6.0 software (GraphPad Software) was used for statistical analysis. The statistical significance of group differences was analyzed with one-way ANOVA followed by Tukey's test or Newman–Kueuls test. P < 0.05 was considered statistically significant.Results
Zey inhibits cell viability and proliferation in K562 cells
MTT assay was performed to evaluate the effect of Zey on the viability of K562 cells. Chemical structure of Zey was shown in Fig. 1A. The cells were treated with various concentrations of Zey for 24 h. As shown in Fig. 1B, Zey treatments reduced the cell viability of the K562 cells in a dose dependent manner with an IC50 of 0.82 μM, indicating that Zey treatment dose-dependently reduced the survival of K562 cells. | ||
| Fig. 1 (A) Chemical structure of zeylenone. (B) Zey suppresses viability of K562 cells. Cell viability of K562 cells treated with Zey was measured using the MTT assay. Cells were seeded in 96-well culture plates, and were treated with Zey at indicated concentrations (0, 0.15, 0.31, 0.625, 1.25, 2.5, 5 and 10 μM) for 24 hours. Data are expressed as means ± SD of 3 distinct experiments. The cell viability of the control (DMSO alone) is indicated as 100%. *P < 0.05 and **P < 0.01 were considered statistically significant. (C) Representative photographs of colony formation for K562 cells treated with increasing concentrations of Zey for 14 days. 1 mg mL−1 MTT solution was added to facilitate colony counting. (D) Colony numbers of K562 cells from three consistent experiments under microscope (middle panel 40×, lower panel 200×). *P < 0.05, **P < 0.01 compared with control untreated cells. | ||
To test the ability of Zey to inhibit anchorage – independent growth of K562 cells, a colony formation assay in soft agar was performed, which was considered one of the best in vitro indicators of tumor malignancy. Representative culture plates from K562 cells were depicted in Fig. 1C, and the number of colonies corresponding to this cell line was shown in Fig. 1D. These data showed that Zey treatment could significantly inhibited colony formation of K562 cells in soft agar as indicated by smaller and fewer colonies compared to untreated control cells.
To further confirm whether Zey inhibits proliferation, we identified the expression of Proliferating Cell Nuclear Antigen (PCNA). Western blot analysis (Fig. 2A) showed that the treatment with Zey significantly decreased the expression of PCNA as compared to the control without Zey treatment. Furthermore, our results by immunofluorescence (Fig. 2B) also confirmed that PCNA level was significantly decreased after cells were treated with Zey for 24 h. These results indicated that proliferation of K562 cells could be significantly inhibited by Zey treatment.
 | ||
| Fig. 2 Zey inhibits proliferation of K562 cells. (A) Western blot analysis of PCNA in K562 cells. K562 cells were dose-dependently treated with Zey for 24 h. Whole-cell lysates were immunoblotted with specific antibodies to PCNA. The data represented the means ± SD for triplicate determinations. **P < 0.01 compared with control untreated cells. (B) Immunofluorescence of PCNA after K562 cells were treated with Zey for 24 h. | ||
Zey induces apoptosis in K562 cells
In order to investigate whether the cytotoxicity was related to apoptosis, K562 cells were treated with Zey for 24 h, and then morphological changes were observed under microscope and photographed. As shown in Fig. 3A, a dose dependent increase of crushed cells could be observed after Zey treatment suggesting that Zey induced cell death in K562 cells in a dose dependent manner. | ||
| Fig. 3 Zey induces apoptosis of K562 cells. (A) Representative images of K562 cell morphology. Cells were treated with Zey at indicated concentrations (0, 0.4, 0.8, and 1.6 μM) for 24 h, and imaged using an Olympus digital camera (magnification 200×). (B) Morphological observation of fluorescence microscope with AO/EB double staining (magnification 100×). | ||
AO/EB staining was simultaneously performed to reveal vigorously growing cells (green) and necrosis or terminal apoptosis cells (orange). The microscopic examination (Fig. 3B) showed that the untreated control K562 cells displayed normal green nuclei, while in Zey-treated cells orange nuclei emerged, and the number of orange nuclei increased in a dose dependent manner, indicating apoptosis after the cells were treated with Zey.
Moreover, apoptosis was determined by flow cytometry, including early apoptotic cells (annexin V-FITC positive, PI negative) and late apoptotic cells (annexin V-FITC and PI positive). After treated with Zey for 24 h, K562 cells were stained with annexin V-FITC/PI. As shown in Fig. 4A and B, Zey induced apoptosis in a dose- and time-dependent manner in K562 cells. The apoptosis percents reached up to 51.15% and 88.16%, respectively, after K562 cells were treated with Zey (1.6 μM) for 24 h and 48 h.
 | ||
| Fig. 4 Zey induces apoptosis in K562 cells detected by the annexin V-FITC/PI staining test. (A) After treated with Zey for 24 h, K562 cells were stained with annexin V-FITC/PI and subjected to flow cytometry (FACScan, Becton Dickinson) to determine apoptotic cells. (B) The ratio of apoptosis cells, including early apoptotic cells (lower right quadrant) and late apoptotic cells (upper right quadrant). The data represented the means ± SD for triplicate determinations. *P < 0.05, **P < 0.01 compared with control untreated cells. | ||
Zey induces the apoptosis in K562 cells via the mitochondrial apoptosis pathway
To understand the underlying mechanism by which Zey induces apoptosis in K562 cells, we investigated changes in the mitochondrial membrane potential and the level of corresponding proteins involved in apoptosis.Initially, we explored the effects of Zey on mitochondrial membrane potential. After treated with Zey for 24 h, cells were stained with JC-1, and the changes of mitochondrial membrane potential were analyzed by flow cytometry. As shown in Fig. 5A, Zey treatment induced significant loss of mitochondrial membrane potential in a dose dependent manner.
 | ||
| Fig. 5 Zey induces apoptosis of K562 cells through the initiation of the mitochondrial pathway. (A) Zey induces decrease of mitochondrial membrane potential in K562 cells. Cells were treated with increasing concentrations of Zey for 24 h, mitochondrial membrane potential were analyzed by flow cytometry after cells were stained with JC-1. The data represented the means ± SD for triplicate determinations. *P < 0.05, **P < 0.01 compared with control untreated cells. (B) Zey induces cleavage of PARP and caspases-3, -8, -9 in K562 cells. Cells were treated with Zey in a dose-dependent manner for 24 h, whole-cell lysates were immunoblotted with specific antibodies to PARP, pro-and cleaved-caspase-3, -8, -9, and β-actin. The data represented the means ± SD for triplicate determinations. *P < 0.05, **P < 0.01 compared with control untreated cells. (C) Zey induces AIF and cytochrome C release. K562 cells were exposed to Zey for 24 h, then cell cytoplasm were extracted by digitonin buffer and the released AIF and cytochrome C were detected by western blot analysis. The data represented the means ± SD for triplicate determinations. *P < 0.05, **P < 0.01 compared with control untreated cells. (D) Zey inhibited expression of antiapoptotic proteins survivin, Bcl-XL, XIAP, and Bcl-2. Cells were treated with Zey in a dose-dependent manner for 24 h, whole-cell lysates were analyzed by western blot analysis. The data represented the means ± SD for triplicate determinations. *P < 0.05, **P < 0.01 compared with control untreated cells. (E) The mRNA levels of survivin, Bcl-XL, XIAP, and Bcl-2 were analyzed by RT-qPCR. Results were normalized according to β-actin levels. The data represented the means ± SD for triplicate determinations. *P < 0.05, **P < 0.01 compared with control untreated cells. | ||
Next, to assess the effects of Zey on the cleavage of caspases and PARP in K562 cells, cells were treated with Zey for 24 h, and then total cell lysates were prepared for western blot analysis. As shown in Fig. 5B, Zey dose-dependently induced the cleavage of PARP which was recognized as an indicator of apoptosis. Consistently, decreased levels of pro-caspases-3 and -8 were detected and that of the cleaved-caspases-3, -8, and -9 were markedly elevated after Zey treatment, paralleled with the pattern of cleaved-PARP. These results indicated that Zey treatment induced apoptosis in K562 cells via caspase-3, caspase-9, and caspase-8 activation. Mitochondria is well known as the regulating center of apoptosis. Release of cytochrome C and AIF from mitochondria to the cytoplasm has been considered as an indicator of apoptosis. Data in Fig. 5C showed that release of cytochrome C and AIF from mitochondria to the cytoplasm increased markedly in a dose-dependent manner after Zey treatment, indicating that mitochondria may involve in Zey-induced apoptosis. To further investigate the mechanism of Zey-induced apoptosis, we measured the effect of Zey on other proteins relate to apoptosis. Western blot analysis showed that Zey treatment induced significant decline of anti-apoptotic proteins Bcl-xl, survivin, XIAP, and Bcl-2 (Fig. 5D). RT-qPCR analysis showed similar results that treatment with Zey significantly decreased mRNA levels of Bcl-xl, survivin, XIAP and Bcl-2 in K562 cells (Fig. 5E), verifying involvement of anti-apoptotic proteins Bcl-xl, survivin, XIAP and Bcl-2 in Zey-induced apoptosis.
Zey downregulates phosphorylation of Jak2 and Src proteins and inhibits their downstream signaling
Jak2 and Src are reported to involved in malignant progress of CML, via activating crucial cellular substrates, including Stat3. Jak2/Stat3 or Src/Stat3 is emerging as a compelling pathway for CML therapy. To assess whether Zey blocks phosphorylation of Jak2, Src and Stat3 in K562 cells derived from CML, western blot analysis was performed with specific antibodies to p-Jak2, p-Src and p-Stat3. The result revealed that Zey treatment resulted in strong reduction of the levels of p-Jak2, p-Src and p-Stat3 (Fig. 6A). Next, to further investigate whether Zey inhibits nuclear translocation of Stat3, nucleus protein was isolated and analyzed by western blot. As expected, consistent with inhibition of phosphorylation of Stat3, Stat3 translocation to nucleus was substantially abrogated (Fig. 6B). Our western blot results were further confirmed by immunofluorescence, showing the similar downregulation of p-Jak2 and p-Src with Zey treatment (Fig. 6C). These results indicated that Zey treatment inhibited phosphorylation of Jak2 and Src in K562 cells, followed by blockade of downstream Stat3 signaling.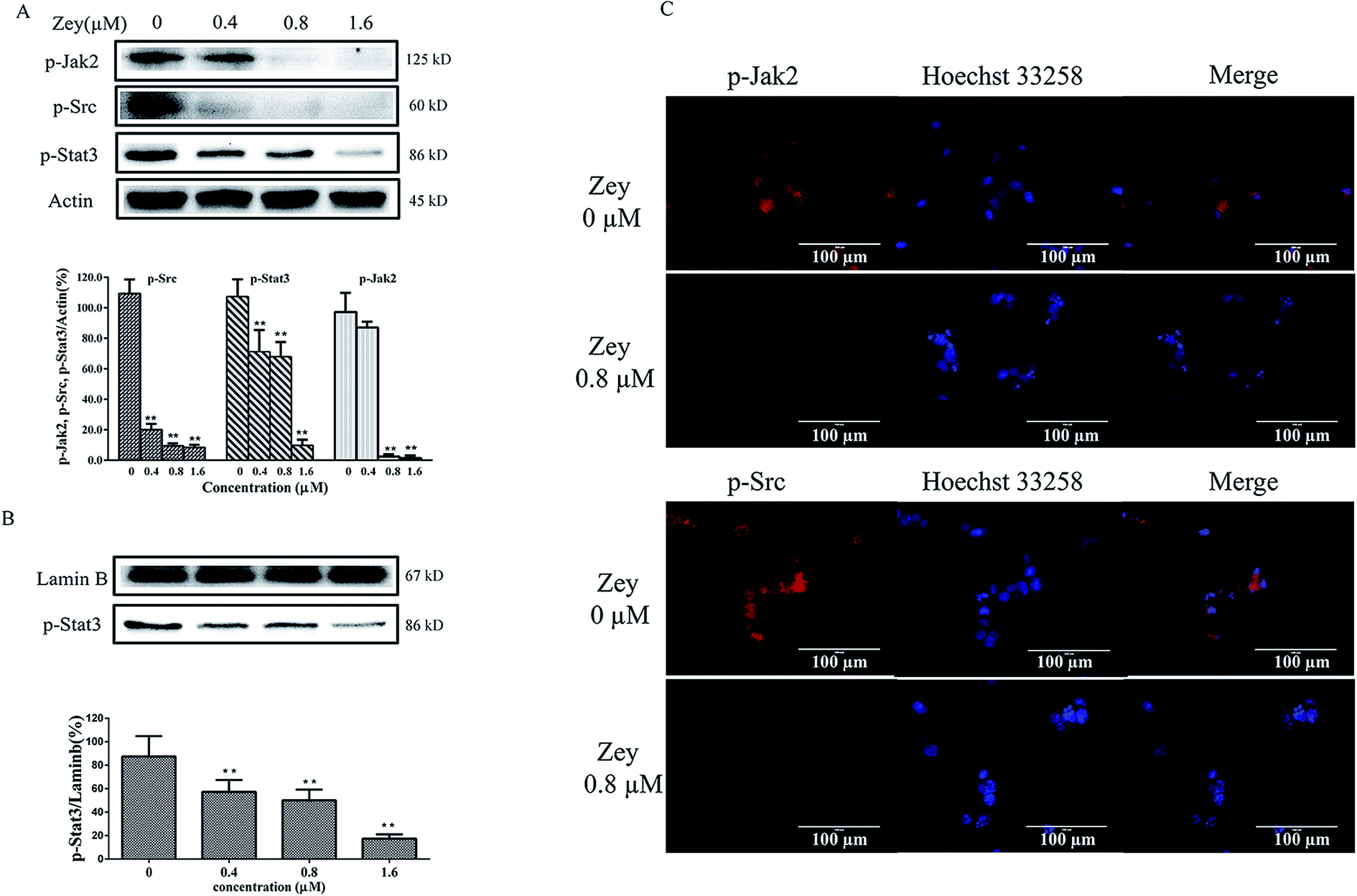 | ||
| Fig. 6 Zey reduces levels of p-Jak2, p-Src and p-Stat3, and inhibits Stat3 nuclear translocation in K562 cells. (A) Western blot analysis of p-Jak2, p-Src and p-Stat3 in K562 cells. K562 cells were dose-dependently treated with Zey for 24 h. Whole-cell lysates were immunoblotted with specific antibodies to p-Jak2, p-Src and p-Stat3. (B) For nuclear translocation analysis, nuclear extracts from K562 cells treated with Zey were prepared and immunoblotted with specific antibodies to p-Stat3. The data represented the means ± SD for triplicate determinations. *P < 0.05, **P < 0.01 compared with control untreated cells. (C) Immunofluorescence of p-Jak2 and p-Src after treatment of Zey in K562 cells for 24 h. | ||
Moreover, many other downstream proteins associated with the proliferation and cell survival including PI-3 kinase, p-ERK and JNK could be influenced by Jak2 and Src, we therefore investigated whether Zey treatment impacted any of these proteins. After exposing K562 cells to Zey for 24 h, expression and phosphorylation of proteins corresponding to PI-3 kinase, p-ERK and JNK were detected by western blot analysis. As shown in Fig. 7A, Zey treatment significantly decreased phosphorylation of p85α subunit of PI3K and its downstream proteins AKT and mTOR in a dose-dependent manner, however, no significant changes were detected in expression of P110 subunit and p-PTEN. Moreover, phosphorylation of p-ERK was also significantly decreased after treatment with Zey, but Zey did not dramatically affect the phosphorylation of JNK compared with the other Jak2 and Src downstream proteins in K562 cells (Fig. 7B).
 | ||
| Fig. 7 Zey down-regulates downstream signaling proteins of Jak2 and Src. (A) and (B) K562 cells were treated with Zey in a dose-dependent manner for 24 h. Whole-cell lysates were immunoblotted with specific antibodies, β-actin was used to confirm equal protein loading. The data represented the means ± SD for triplicate determinations. *P < 0.05, **P < 0.01 compared with control untreated cells. | ||
Zey suppresses tumor growth in a mouse xenograft model
To verify our findings from in vitro studies and demonstrate the anticancer activity of Zey, the in vivo study was carried out with a K562 cell tumor xenograft mouse model, followed by treatments with mPEG-PLGA loaded zeylenone micelles (30 mg kg−1) and IM (50 mg kg−1) by intraperitoneal injection once a day for 14 days. As shown in Fig. 8A, Zey treatment significantly suppressed the tumor growth as compared with the control group. Isolated tumor weight was also remarkably reduced in the Zey treated group than that in the control group (Fig. 8B). Moreover, mouse body weight remained relatively stable in each group and no side effects were observed in Zey-treated mice (Fig. 8C). | ||
| Fig. 8 Zey inhibits tumor growth, and reduces levels of p-Jak2, p-Src and p-Stat3 in nude BALB/c mice bearing K562 xenograft. Nude mice bearing K562 xenograft tumors were treated with vehicle, Imatinib (50 mg per kg per day, i.p.), mPEG-PLGA loaded zeylenone micelles (30 mg per kg per day, i.p.) for 14 days. (A) Tumor volume were recorded every 2 days. Mean ± SD (n = 5). *P < 0.05, **P < 0.01, versus control group. (B) Tumor weight on day 14. After the mice were sacrificed, the tumor tissues were isolated, weighed, and summarized. *P < 0.05, **P < 0.01, versus control group. (C) Mouse body weight was measured every 2 days. Mean ± SD (n = 5). (D) Expression of p-Jak2, p-Src and p-Stat3 in tumor tissues was detected by Immunohistochemical analysis (magnification 400×). | ||
An immunohistochemical (IHC) analysis of the xenograft tumor tissues was performed to further confirm whether the underlying mechanism was associated with downregulating phosphorylation of Jak2, Src and Stat3 proteins. As expected, treatment with Zey decreased the levels of p-Jak2, p-Src and p-Stat3 as compared to the control group (Fig. 8D). Collectively, these results demonstrated the anticancer activity of Zey in vivo against K562 cells in a mouse xenograft model.
To further expore toxicity of Zey in vivo, H&E staining of kidney, spleen, and liver from Zey-treated and untreated nude mice bearing K562 xenograft tumors was performed. As shown in Fig. 9, no signs of toxicity were observed in Zey-untreated mice or mice that received 30 mg kg−1 Zey for 14 days.
 | ||
| Fig. 9 H&E staining of sections from kidney, spleen, and liver of Zey-treated and untreated nude mice bearing K562 xenograft tumors (magnification 400×). After the mice were sacrificed, kidney, spleen, and liver were isolated and fixed in 10% phosphate-buffered formalin for H&E staining. | ||
Discussion
Previous studies indicate that Zey inhibits proliferation and induces apoptosis in a variety of cancer cells, including cervical, breast, lung, prostate carcinoma, and acute lymphoblastic leukemia.17 We, therefore, proposed that Zey may be a potent compound in suppression of CML cell line K562. In the present study, we evaluated the anticancer effects of Zey in K562 cells both in vitro and in a mouse xenograft model, and further investigated the underlying mechanisms. To the best of our knowledge, this study for the first time demonstrated that Zey treatment inhibited cell viability, suppressed colony formation and increased apoptosis in K562 cells through inducing the mitochondrial-mediated apoptosis pathway, downregulating phosphorylation of Jak2 and Src proteins and thus inhibiting their downstream signaling. Besides, Zey showed significantly anticancer activity in K562 xenografts, better than that of IM, a paradigm for targeted therapy for CML. These findings suggest that the natural herbal product derivative Zey is a promising therapeutic agent for CML treatment, distinguished from BCR–ABL inhibitor, through suppressing phosphorylation of Jak2 and Src in K562 cells.Previous studies have confirmed that the imbalance between apoptotic proteins such as Bax versus antiapoptotic factors such as Bcl-2, XIAP, and survivin would result in decrease of mitochondrial membrane integrity, thus leading to the release of cytochrome C and AIF from mitochondria to the cytoplasm. The released cytochrome C then activated caspase either directly or by forming caspase-9 complex, after which it induced PARP cleavage and then apoptosis.23–25 Consistently, in this study, we found that Zey treatment induced apoptosis by dose-dependently decreased levels of antiapoptotic proteins including Bcl-2, Bcl-XL, survivin and XIAP, and significantly induced loss of mitochondrial membrane potential, release of cytochrome C and AIF, and activation of mitochondrial downstream caspases and the onset of apoptosis. Moreover, pro-caspase-8 was cleaved and its total level was decreased in a dose dependent manner with treatment of Zey, indicating that the receptor mediated death pathways may be involved in Zey-induced apoptosis.
STAT proteins play essential roles in normal cytokine signaling and are aberrantly activated in human cancers.26 Particularly, Stat3, a member of STAT family activated by cytokines and tyrosine kinases, could transmit signals from the cell surface to the nucleus to regulate transcription of target genes (e.g., Bcl-XL, Bcl-2).27,28 The activation of Stat3 protein is a rapid and transient event that contributes to tumor growth by initiating the cell cycle, preventing apoptosis, and up-regulating oncogenes. Several studies have highlighted that blockade of persistent Stat3 could inhibit cancer cell survival both in vitro and in vivo.29,30 Therefore, inhibition of Stat3 signaling by a direct or indirect method, might be considered profitable tools to treat CML disease. In the present study, our data demonstrated that Zey could significantly reduce phosphorylation of Stat3, and thereby inhibiting its nuclear relocation, indicating consistent de-phosphorylation of Stat3 by Zey treatment.
However, it remains unclear that whether Stat3 inhibition by Zey treatment is associated with suppression of the upstream kinases. JAK and Src are reported as two major tyrosine kinases upstream of Stat3,31 thus appealing much of our attention. JAKs, are a family of non-receptor tyrosine kinases, comprising four members with similar structures and functions shared among each of them, namely Jak1, Jak2, Jak3 and Tyk2,32,33 among which, Jak2 is most frequently activated in CML.34 Different cytokines and growth factors may result in activation of Jak2, thereby phosphorylating STAT family members, including Stat3, leading to their nuclear relocation.35–37 Jak2/Stat3 signaling is tightly regulated in normal cells, however, it is persistently activated in cancer cells due to the aberrant activation of Jak2 or other tyrosine kinases.38 As one of the more recently recognized oncogenic signaling pathways, Jak2/Stat3 signaling has been comprehensively examined in haematological malignancies and considered as a promising target in the treatment of CML. Therefore, we identified whether Zey could inhibit Jak2/Stat3 signaling pathway. As expected, in the current study, we found that Zey significantly inhibited phosphorylation of Jak2 both in vivo and in vitro. Our data indicate that Zey inhibited Stat3 phosphorylation through inhibiting the phosphorylation of the upstream kinases Jak2.
Src, also nonreceptor tyrosine kinase, is another upstream regulator of Stat3.39 It participates in several normal cellular functions, including cell cycle progression, apoptosis regulation, cell survival, and differentiation.40,41 However, in multiple cancer cell types, Src is aberrantly activated, thus deregulating downstream proteins, via phosphorylating crucial cellular substrates on tyrosyl residues, including STAT family members, especially Stat3.42,43 Inhibition of Src phosphorylation and the subsequent oncogenic signaling cascades using both pharmacologic inhibitors and molecular approaches in a variety of human cancer cell lines and patient tumors has been found to be very effective in suppression of proliferation, cell cycle arrest, and inhibition of tumor growth in vivo,44,45 such as dasatinib, a SFK inhibitor, which was approved for leukemia treatment in 2006, providing scientific rationale for the continued investigation of SFK inhibitor as a therapeutic strategy for patients suffering from CML disease. In this study, our results by western blot, immunofluorescence and IHC analysis demonstrated that Zey significantly inhibited phosphorylation of Src both in vivo and in vitro.
Moreover, JAKs and SFKs could cooperate to constitutively activate Stat3 signaling.18 Previous studies have demonstrated the compensatory effect of Jak2 in response to prolonged inhibition of Src in cancer,31 when Src was inhibited in cancer cells by a Src inhibitor, phosphorylation of Src was decreased, however, to compensate Src inhibition phosphorylation of Jak2 was still maintained. Therefore, suppression of Stat3 was less effective when treated with a Src inhibitor alone.28 In this study, we found that Zey, with potent anticancer activity in CML cell lines and in a mouse xenograft model, could overcome this compensatory drawback through blockade of both Jak2 and Src phosphorylation, which may be more effective than a Jak or Src inhibitor.
Besides Stat3, aberrant activation of the Jak2 and Src enhances a number of other oncogenic signaling molecules and pathways including PI3K/Akt/mTOR and Ras/Raf/MEK/MAPK (ERK1/2),46,47 which are associated with the progression of haematological malignancies, providing a basis for their investigation in CML. Here, we demonstrate that Zey could inhibit tumor development via inhibiting phosphorylation of the p85α subunit of PI3K and thus the downstream proteins AKT and mTOR, moreover, phosphorylation of ERK1/2 was also decreased after the treatment with Zey in K562 cells.
In summary, we report that the natural product Zey could inhibit phosphorylation of Jak2 and Src in K562 cells, which subsequently inhibits Stat3, PI3K/AKT, and ERK1/2 signaling pathways (Fig. 10), though the potential mechanism of the reduction of Jak2 and Src is unclear and we cannot exclude other molecular targets of Zey involved in this cell line. In addition, our results clearly demonstrated that Zey markedly inhibited K562 xenografts through inhibiting phosphorylation of Jak2, Src and Stat3. Thus, Zey is promising for development as a novel therapeutic agent targeting Jak2 and Src signaling in CML.
 | ||
| Fig. 10 A schematic illustration of a proposed mechanism by which Zey induces cytotoxicity in K562 cells. | ||
Conflict of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by PUMC Youth Fund (No. 3332015047) and the Fundamental Research Funds for the Central Universities, and by CAMS Innovation Fund for Medical Sciences (CIFMS) (No. 2016-I2M-1-012). This work was also supported by Key Laboratory of Bioactive Substances and Resources Utilization of Chinese Herbal Medicine, Ministry of Education, and by Beijing Key Laboratory of Innovative Drug Discovery of Traditional Chinese Medicine (Natural Medicine) and Translational Medicine, Institute of Medical Plant Development, Peking Union Medical College and Chinese Academy of Medical Sciences.References
- W. Tao, S. N. Chakraborty, X. Leng, H. Ma and R. B. Arlinghaus, HSP90 inhibitor AUY922 induces cell death by disruption of the Bcr–Abl, Jak2 and HSP90 signaling network complex in leukemia cells, Genes Cancer, 2015, 6, 19–29 Search PubMed.
- A. Tefferi and D. G. Gilliland, Oncogenes in myeloproliferative disorders, Cell Cycle, 2007, 6, 550–566 CrossRef CAS PubMed.
- H. Modi, L. Li, S. Chu, J. Rossi, J. K. Yee and R. Bhatia, Inhibition of Grb2 expression demonstrates an important role in BCR–ABL-mediated MAPK activation and transformation of primary human hematopoietic cells, Leukemia, 2011, 25, 305–312 CrossRef CAS PubMed.
- Z. L. Huang, M. Gao, Q. Y. Li, K. Tao, Q. Xiao, W. X. Cao and W. L. Feng, Induction of apoptosis by directing oncogenic Bcr–Abl into the nucleus, Oncotarget, 2013, 4, 2249–2260 CrossRef PubMed.
- A. Quintas-Cardama and J. Cortes, Molecular biology of bcr–abl1-positive chronic myeloid leukemia, Blood, 2009, 113, 1619–1630 CrossRef CAS PubMed.
- T. G. Lugo, A. M. Pendergast, A. J. Muller and O. N. Witte, Tyrosine kinase activity and transformation potency of bcr–abl oncogene products, Science, 1990, 247, 1079–1082 CAS.
- E. K. Thomas, J. A. Cancelas, Y. Zheng and D. A. Williams, Rac GTPases as key regulators of p210-BCR–ABL-dependent leukemogenesis, Leukemia, 2008, 22, 898–904 CrossRef CAS PubMed.
- M. Savona and M. Talpaz, Chronic myeloid leukemia: changing the treatment paradigms, Oncology, 2006, 20, 707–711 Search PubMed ; discussion 712-704, 719, 724.
- A. Hochhaus, B. Druker, C. Sawyers, F. Guilhot, C. A. Schiffer, J. Cortes, D. W. Niederwieser, C. Gambacorti-Passerini, R. M. Stone and J. Goldman, et al., Favorable long-term follow-up results over 6 years for response, survival, and safety with imatinib mesylate therapy in chronic-phase chronic myeloid leukemia after failure of interferon-alpha treatment, Blood, 2008, 111, 1039–1043 CrossRef CAS PubMed.
- B. J. Druker, F. Guilhot, S. G. O'Brien, I. Gathmann, H. Kantarjian, N. Gattermann, M. W. Deininger, R. T. Silver, J. M. Goldman and R. M. Stone, et al., Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia, N. Engl. J. Med., 2006, 355, 2408–2417 CrossRef CAS PubMed.
- H. Yang, H. Hui, Q. Wang, H. Li, K. Zhao, Y. Zhou, Y. Zhu, X. Wang, Q. You and Q. Guo, et al., Wogonin induces cell cycle arrest and erythroid differentiation in imatinib-resistant K562 cells and primary CML cells, Oncotarget, 2014, 5, 8188–8201 CrossRef PubMed.
- P. Neviani, R. Santhanam, J. J. Oaks, A. M. Eiring, M. Notari, B. W. Blaser, S. Liu, R. Trotta, N. Muthusamy and C. Gambacorti-Passerini, et al., FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia, J. Clin. Invest., 2007, 117, 2408–2421 CAS.
- A. Quintas-Cardama, H. Kantarjian and J. Cortes, Flying under the radar: the new wave of BCR–ABL inhibitors, Nat. Rev. Drug Discovery, 2007, 6, 834–848 CrossRef CAS PubMed.
- F. Grebien, O. Hantschel, J. Wojcik, I. Kaupe, B. Kovacic, A. M. Wyrzucki, G. D. Gish, S. Cerny-Reiterer, A. Koide and H. Beug, et al., Targeting the SH2-kinase interface in Bcr–Abl inhibits leukemogenesis, Cell, 2011, 147, 306–319 CrossRef CAS PubMed.
- E. Jabbour, A. Hochhaus, J. Cortes, P. La Rosee and H. M. Kantarjian, Choosing the best treatment strategy for chronic myeloid leukemia patients resistant to imatinib: weighing the efficacy and safety of individual drugs with BCR–ABL mutations and patient history, Leukemia, 2010, 24, 6–12 CrossRef CAS PubMed.
- M. A. Macha, S. Rachagani, S. Gupta, P. Pai, M. P. Ponnusamy, S. K. Batra and M. Jain, Guggulsterone decreases proliferation and metastatic behavior of pancreatic cancer cells by modulating JAK/STAT and Src/FAK signaling, Cancer Lett., 2013, 341, 166–177 CrossRef CAS PubMed.
- L. Zhang, J. Jin, L. Zhang, R. Hu, L. Gao, X. Huo, D. Liu, X. Ma, C. Wang and J. Han, et al., Quantitative analysis of differential protein expression in cervical carcinoma cells after zeylenone treatment by stable isotope labeling with amino acids in cell culture, J. Proteomics, 2015, 126, 279–287 CrossRef CAS PubMed.
- L. H. Sun, H. X. Dang, L. L. Piao, Y. H. Liao, Y. H. Yu, X. M. Liu, C. E. Cox, R. Falcone, R. Fairclough and S. Parsons, et al., Effects of zeylenone on the proliferation and apoptosis of acute lymphoblastic leukemia cells, J. Tradit. Chin. Med., Univ. Hunan, 2012, 32, 15–20 CAS.
- X. Hu, R. Han, L. H. Quan, C. Y. Liu and Y. H. Liao, Stabilization and sustained release of zeylenone, a soft cytotoxic drug, within polymeric micelles for local antitumor drug delivery, Int. J. Pharm., 2013, 450, 331–337 CrossRef CAS PubMed.
- Y. Wang, J. Chen, L. Wang, Y. Huang, Y. Leng and G. Wang, Fangchinoline induces G0/G1 arrest by modulating the expression of CDKN1A and CCND2 in K562 human chronic myelogenous leukemia cells, Exp. Ther. Med., 2013, 5, 1105–1112 CAS.
- H. Yang, H. R. Lawrence, A. Kazi, H. Gevariya, R. Patel, Y. Luo, U. Rix, E. Schonbrunn, N. J. Lawrence and S. M. Sebti, Dual Aurora A and JAK2 kinase blockade effectively suppresses malignant transformation, Oncotarget, 2014, 5, 2947–2961 CrossRef PubMed.
- S. J. Kim, K. H. Jung, H. H. Yan, M. K. Son, Z. Fang, Y. L. Ryu, H. Lee, J. H. Lim, J. K. Suh and J. Kim, et al., HS-543 induces apoptosis of Imatinib-resistant chronic myelogenous leukemia with T315I mutation, Oncotarget, 2015, 6, 1507–1518 CrossRef PubMed.
- A. F. Nica, C. C. Tsao, J. C. Watt, T. Jiffar, S. Kurinna, P. Jurasz, M. Konopleva, M. Andreeff, M. W. Radomski and P. P. Ruvolo, Ceramide promotes apoptosis in chronic myelogenous leukemia-derived K562 cells by a mechanism involving caspase-8 and JNK, Cell Cycle, 2008, 7(21), 3362–3370 CrossRef CAS PubMed.
- R. C. Taylor, S. P. Cullen and S. J. Martin, Apoptosis: controlled demolition at the cellular level, Nat. Rev. Mol. Cell Biol., 2008, 9, 231–241 CrossRef CAS PubMed.
- S. H. Kaufmann and M. O. Hengartner, Programmed cell death: alive and well in the new millennium, Trends Cell Biol., 2001, 11, 526–534 CrossRef CAS PubMed.
- S. Nam, A. Scuto, F. Yang, W. Chen, S. Park, H. S. Yoo, H. Konig, R. Bhatia, X. Cheng and K. H. Merz, et al., Indirubin derivatives induce apoptosis of chronic myelogenous leukemia cells involving inhibition of Stat5 signaling, Mol. Oncol., 2012, 6, 276–283 CrossRef CAS PubMed.
- S. J. Thomas, J. A. Snowden, M. P. Zeidler and S. J. Danson, The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours, Br. J. Cancer, 2015, 113, 365–371 CrossRef CAS PubMed.
- B. Sen, B. Saigal, N. Parikh, G. Gallick and F. M. Johnson, Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding, Cancer Res., 2009, 69, 1958–1965 CrossRef CAS PubMed.
- M. A. Blaskovich, J. Sun, A. Cantor, J. Turkson, R. Jove and S. M. Sebti, Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice, Cancer Res., 2003, 63, 1270–1279 CAS.
- J. Sun, M. A. Blaskovich, R. Jove, S. K. Livingston, D. Coppola and S. M. Sebti, Cucurbitacin Q: a selective STAT3 activation inhibitor with potent antitumor activity, Oncogene, 2005, 24, 3236–3245 CrossRef CAS PubMed.
- L. Liu, N. Gaboriaud, K. Vougogianopoulou, Y. Tian, J. Wu, W. Wen, L. Skaltsounis and R. Jove, MLS-2384, a new 6-bromoindirubin derivative with dual JAK/Src kinase inhibitory activity, suppresses growth of diverse cancer cells, Cancer Biol. Ther., 2014, 15, 178–184 CrossRef CAS PubMed.
- J. J. Babon, I. S. Lucet, J. M. Murphy, N. A. Nicola and L. N. Varghese, The molecular regulation of Janus kinase (JAK) activation, Biochem. J., 2014, 462, 1–13 CrossRef CAS PubMed.
- K. Yamaoka, P. Saharinen, M. Pesu, V. E. Holt 3rd, O. Silvennoinen and J. J. O'Shea, The Janus kinases (Jaks), Genome Biol., 2004, 5, 253 CrossRef PubMed.
- A. K. Samanta, S. N. Chakraborty, Y. Wang, E. Schlette, E. P. Reddy and R. B. Arlinghaus, Destabilization of Bcr-Abl/Jak2 Network by a Jak2/Abl Kinase Inhibitor ON044580 Overcomes Drug Resistance in Blast Crisis Chronic Myelogenous Leukemia (CML), Genes Cancer, 2010, 1, 346–359 CrossRef CAS PubMed.
- S. Nam, W. Wen, A. Schroeder, A. Herrmann, H. Yu, X. Cheng, K. H. Merz, G. Eisenbrand, H. Li and Y. C. Yuan, et al., Dual inhibition of Janus and Src family kinases by novel indirubin derivative blocks constitutively-activated Stat3 signaling associated with apoptosis of human pancreatic cancer cells, Mol. Oncol., 2013, 7, 369–378 CrossRef CAS PubMed.
- T. Kisseleva, S. Bhattacharya, J. Braunstein and C. W. Schindler, Signaling through the JAK/STAT pathway, recent advances and future challenges, Gene, 2002, 285, 1–24 CrossRef CAS PubMed.
- D. S. Aaronson and C. M. Horvath, A road map for those who don't know JAK-STAT, Science, 2002, 296, 1653–1655 CrossRef CAS PubMed.
- L. Liu, S. Nam, Y. Tian, F. Yang, J. Wu, Y. Wang, A. Scuto, P. Polychronopoulos, P. Magiatis and L. Skaltsounis, et al., 6-Bromoindirubin-3′-oxime inhibits JAK/STAT3 signaling and induces apoptosis of human melanoma cells, Cancer Res., 2011, 71, 3972–3979 CrossRef CAS PubMed.
- L. A. Byers, B. Sen, B. Saigal, L. Diao, J. Wang, M. Nanjundan, T. Cascone, G. B. Mills, J. V. Heymach and F. M. Johnson, Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer, Clin. Cancer Res., 2009, 15, 6852–6861 CrossRef CAS PubMed.
- F. M. Johnson and G. E. Gallick, SRC family nonreceptor tyrosine kinases as molecular targets for cancer therapy, Adv. Anticancer Agents Med. Chem., 2007, 7, 651–659 CrossRef CAS.
- T. J. Yeatman, A renaissance for SRC, Nat. Rev. Cancer, 2004, 4, 470–480 CrossRef CAS PubMed.
- S. J. Parsons and J. T. Parsons, Src family kinases, key regulators of signal transduction, Oncogene, 2004, 23, 7906–7909 CrossRef CAS PubMed.
- H. Yu and R. Jove, The STATs of cancer–new molecular targets come of age, Nat. Rev. Cancer, 2004, 4, 97–105 CrossRef CAS PubMed.
- G. Niu, T. Bowman, M. Huang, S. Shivers, D. Reintgen, A. Daud, A. Chang, A. Kraker, R. Jove and H. Yu, Roles of activated Src and Stat3 signaling in melanoma tumor cell growth, Oncogene, 2002, 21, 7001–7010 CrossRef CAS PubMed.
- J. G. Trevino, J. M. Summy, D. P. Lesslie, N. U. Parikh, D. S. Hong, F. Y. Lee, N. J. Donato, J. L. Abbruzzese, C. H. Baker and G. E. Gallick, Inhibition of SRC expression and activity inhibits tumor progression and metastasis of human pancreatic adenocarcinoma cells in an orthotopic nude mouse model, Am. J. Pathol., 2006, 168, 962–972 CrossRef CAS PubMed.
- T. I. Mughal, S. Girnius, S. T. Rosen, S. Kumar, A. Wiestner, O. Abdel-Wahab, J. J. Kiladjian, W. H. Wilson and R. A. Van Etten, Emerging therapeutic paradigms to target the dysregulated Janus kinase/signal transducer and activator of transcription pathway in hematological malignancies, Leuk. Lymphoma, 2014, 55, 1968–1979 CrossRef PubMed.
- T. C. Windham, N. U. Parikh, D. R. Siwak, J. M. Summy, D. J. McConkey, A. J. Kraker and G. E. Gallick, Src activation regulates anoikis in human colon tumor cell lines, Oncogene, 2002, 21, 7797–7807 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra23443g |
| This journal is © The Royal Society of Chemistry 2016 |