
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the purported “backbone fluorescence” in protein three-dimensional fluorescence spectra
Annalisa
Bortolotti†
a,
Yin How
Wong†
b,
Stine S.
Korsholm‡
c,
Noor Hafizan B.
Bahring
b,
Sara
Bobone§
a,
Saad
Tayyab
 *b,
Marco
van de Weert
*c and
Lorenzo
Stella
*a
*b,
Marco
van de Weert
*c and
Lorenzo
Stella
*a
aDipartimento di Scienze e Tecnologie Chimiche, Università di Roma Tor Vergata, 00133 Rome, Italy. E-mail: stella@stc.uniroma2.it; Fax: +39 0672594463; Tel: +39 0672594463
bBiomolecular Research Group, Biochemistry Program, Institute of Biological Sciences, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia. E-mail: saadtayyab2004@yahoo.com; Fax: +603 7967 4178; Tel: +603 7967 7118
cDepartment of Pharmacy, Faculty of Health and Medical Sciences, University of Copenhagen, Universitetsparken 2, 2100 Copenhagen, Denmark. E-mail: marco.vandeweert@sund.ku.dk; Fax: +45 35336030; Tel: +45 35336186
First published on 23rd November 2016
Abstract
In this study, several proteins (albumin, lysozyme, insulin) and model compounds (Trp, Tyr, homopolypeptides) were used to demonstrate the origin of the fluorescence observed upon their excitation at 220–230 nm. In the last 10 years we have observed a worrying increase in the number of articles claiming that this fluorescence originates from the protein backbone, contrary to the established knowledge that UV protein emission is due to aromatic amino acids only. Overall, our data clearly demonstrate that the observed emission upon excitation at 220–230 nm is due to the excitation of Tyr and/or Trp, with subsequent emission from the lowest excited state (i.e. the same as obtained with 280 nm excitation) in agreement with Kasha's rule. Therefore, this fluorescence peak does not provide any information on backbone conformation, but simply reports on the local environment around the aromatic side chains, just as any traditional protein emission spectrum. The many papers in reputable journals erroneously reporting this peak assignment, contradicting 5 decades of prior knowledge, have led to the creation of a new dogma, where many authors and reviewers now take the purported backbone fluorescence as an established fact. We hope the current paper helps counter this new situation and leads to a reassessment of those papers that make this erroneous claim.
Introduction
Fluorescence spectroscopy is a commonly used and powerful method to study protein conformational changes, by taking advantage of the emission properties of aromatic residues, in particular tryptophan (Trp).1,2 Among many different fluorescence approaches, three-dimensional (3D) fluorescence spectra, also called excitation–emission matrices or total fluorescence, represent a powerful tool to analyse complex samples. In this technique, emission intensity is measured for all possible combinations of excitation and emission wavelengths, thus summarizing in a single 3D plot the information corresponding to many different excitation and emission spectra.3–7 This approach is very useful in analysing samples containing multiple fluorophores with overlapping spectra, such as wastewaters.8In a large number of papers, primarily published in the last 10 years, 3D fluorescence spectra have also been used to characterize ligand–protein interactions, and specifically the purported conformational changes induced by the association. Among other components, 3D fluorescence spectra of proteins usually exhibit a peak with an excitation maximum at 220–230 nm, and an emission maximum at 300–350 nm, the exact peak position depending on the specific protein and the fluorimeter used (see Fig. 1 for selected examples of human serum albumin (HSA), lysozyme and human insulin). Surprisingly, in a large number of recent papers this peak has been alleged to originate from the protein backbone and its properties (intensity and position) to be sensitive to conformational changes of that backbone. For example, an analysis of 176 articles published in 2012 and dealing with fluorescence studies of ligand binding to albumin revealed that 24 of those, published in 14 different journals, assigned a peak in 3D fluorescence spectra to backbone emission.9 Also in 2016 many examples of papers making the same claim can be found in the literature.10–26 These latter references are solely meant as examples of the widespread nature of the backbone fluorescence claim and are not an all-inclusive list. More importantly, we would like to point out that 3D fluorescence spectra are simply one of the many experiments reported in those articles. Here we wish to comment only on the use of this particular technique. It should also be noted that some other reports describe self-assembly induced emission in the visible region by peptides in fibrillar or other aggregated states. The still-debated origin of this phenomenon has been recently attributed to proton transfer through intermolecular hydrogen bonds, which could take place only in very specific structures.27 However, here we will limit our discussion to the claim that all protein backbones emit in the UV region.
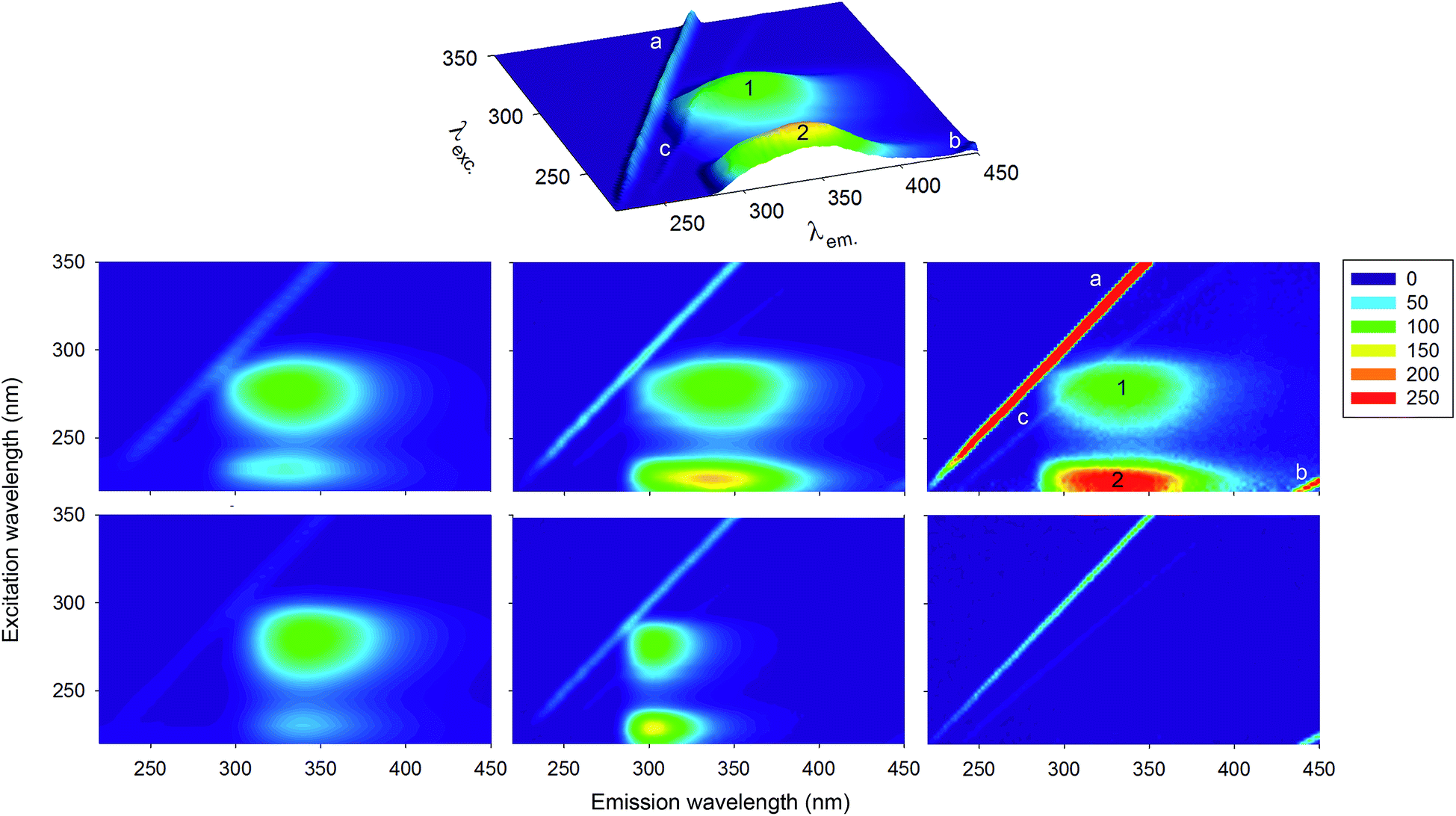 | ||
| Fig. 1 Normalised 3D fluorescence spectra/contour maps of different proteins and of a buffer solution. Top row: 3D fluorescence spectrum of HSA (DK). Middle row: contour maps for HSA collected under different experimental conditions and using different instruments (see Materials and methods) in MY (left), DK (centre) and IT (right). Bottom row: lysozyme (MY, left), insulin (DK, centre) and buffer (IT, right). The peak labels are described in the main text. Fluorescence intensities of all spectra were normalised to 100 on peak ‘1’, with the exception of the buffer spectrum, normalised on the maximum of the ‘a’ band. The colour code for relative fluorescence intensity values is reported on the right. | ||
This claim is actually extremely surprising, as it contradicts half a century of prior knowledge. Already in 1952, Debye and Edwards demonstrated that protein phosphorescence is due to the aromatic residues only.28 In 1953 Gregorio Weber postulated, on the basis of general considerations of the properties of aromatic amino acids and the positions of their absorption maxima, that protein fluorescence originates from aromatic amino acids, too.29 This hypothesis was shortly afterwards confirmed by ground-breaking experimental studies.30–32 In 1967,32 Konev summarized the pioneering years of protein fluorescence studies as follows: “The exclusively aromatic nature of protein fluorescence was conclusively shown by experiments in which no fluorescence could be detected in proteins which contain no aromatic amino acids… It can now be regarded as established that the formation of fluorescence and phosphorescence spectra of proteins involves only three aromatic amino acids which are capable of luminescence in the free state. These are tryptophan, tyrosine, and phenylalanine”.
Also several more recent papers discuss fluorescence obtained by far-UV excitation (220–230 nm range) in the context of excitation and emission by the Tyr and Trp residues.33–35 Thus, in a historical perspective the observation of fluorescence from the protein backbone is a potentially exciting new finding. Unfortunately, an examination of the references used to substantiate this claim reveals no study that directly investigated this phenomenon. If a reference is provided at all, it is to papers that make the same claim, or to papers reporting that the protein backbone absorbs UV-radiation in a broad range between 200 and 250 nm. However, also the aromatic amino acids absorb in this range. We tried to find the original article where backbone emission was first proposed, and identified a work by Zhang et al.36 as a likely first. Here, the authors speculated that, since the backbone absorbs in the far UV region, then the 3D peak “may mainly exhibit the fluorescence characteristic of polypeptide backbone structures”. The only data reported to support this hypothesis was the decrease in peak intensity with increasing protein concentration. This finding, which is in reality likely arising from inner-filter effects,37 was interpreted as the result of the interaction between peptide chains. One of the authors on the current paper was also misled by the aforementioned conclusion in peak assignment.38 It is rather remarkable that the large number of studies using 3D fluorescence spectra to study structural changes of proteins upon ligand binding are based on such insubstantial evidence, against a large historical body of studies stating the opposite.
There are a number of further reasons that cast doubt on the possibility of backbone emission:
(1) Very few molecules are measurably fluorescent, due to the fact that non-radiative decay processes usually predominate over the emission of a photon in the relaxation of excited molecules. Although it is still difficult to predict theoretically which molecules will exhibit fluorescence,39 fluorescent compounds are aromatic, or (less often) highly unsaturated aliphatic molecules. Delocalized electrons, formally present in conjugated double bonds, are required for providing the rigidity necessary to inhibit non-radiative relaxation processes.40–42 No extended conjugated systems are present in the peptide backbone and the low wavelength region of peptide absorption at around 220 nm is due to an n → π* transition,43 which is forbidden in the electric dipole approximation. Both these points argue against the backbone showing any fluorescence.40–42
(2) In their seminal 1957 article on the origin of protein fluorescence, Teale and Weber30 indicated that an important criterion to verify “whether the fluorescence shown by a solution is due to a given substance present in it” is a good match between the fluorescence excitation spectrum and the absorption of the putative fluorophore. By contrast, the protein backbone has an absorption maximum around 190 nm, corresponding to a π → π* transition of the amide band,44 and therefore does not match with the 220–230 nm excitation peak observed in 3D fluorescence spectra.
(3) Fluorescence is always observed at wavelengths longer than those of the lowest energy absorption band. This phenomenon, called Stokes shift,45 is largely due to differential solvation of the excited and ground states (in addition to vibrational relaxation phenomena). Therefore, the solvent-induced shifts of emission bands can be used to calculate dipole moments of electronically excited molecules.46 An absorption–emission shift from 220 to 340 nm would correspond to a Stokes shift of approximately 2 eV in energy. This shift would be much larger than the record shifts reported for charge transfer compounds such as 6-propionyl-2-(N,N-dimethylamino)naphthalene (PRODAN)47 or 4-dimethylamino-4′-nitrostilbene (DANS),48 which undergo an exceptional dipole increase following excitation.
For all the reasons explained above, it should be clear that the peak 220–230/300–350 nm peak observed in 3D fluorescence spectra of proteins cannot be due to backbone emission. To make this point clear from an experimental point of view, we collected 3D fluorescence spectra for different proteins and model compounds. These data will allow us to fully clarify all the features commonly observed in 3D fluorescence spectra of proteins. Samples were analysed independently in different laboratories around the world (Denmark, Italy, and Malaysia). Experiments were performed on different fluorimeters and using different settings, to show the robustness of the observations and of the conclusions drawn, and to illustrate the differences that can be observed in 3D fluorescence spectra due to different experimental settings and instrumentation.
Materials and methods
In this section, differences between the three groups are indicated by the two letter code of the relevant country (DK, IT, MY).Materials
Human serum albumin (HSA), lysozyme from chicken egg white, L-tryptophan (Trp), L-tyrosine (Tyr), poly-DL-alanine (Poly-A), recombinant human insulin, poly-L-lysine (Poly-K), bicinchoninic acid (BCA) kit for protein determination and sodium hydrogen phosphate were purchased from Sigma Aldrich (USA). Octa-L-arginine was from GenScript Corp. (Piscataway, NJ). Sodium dihydrogen phosphate was obtained from VWR (Leuven, Belgium) (DK) or Sigma Aldrich (USA) (IT).UV spectroscopy
UV-visible spectra were acquired using: a V-770 (Jasco) (IT), a Nanodrop 2000C (Thermo Scientific) (DK) or a UV-2450 (Shimadzu) (MY) spectrophotometer.Sample preparation
All samples (IT, DK) were dissolved in sodium phosphate buffer (10 mM, pH 7.4), except the insulin solution that was prepared by first dissolving 5 mg in 80 μl of 0.1 M HCl, then neutralized using 80 μl of 0.1 M NaOH and then diluted using ca. 5 ml of phosphate buffer. This sample was then filtered through a 0.22 μm filter. All samples (MY) were prepared in Milli-Q water except for Tyr and Trp, which were dissolved in 1 M HCl, diluted with Milli-Q water and neutralized with NaOH.The concentrations of the samples containing aromatic moieties were determined spectrophotometrically using molar extinction coefficients of 5630 cm−1 M−1 at 280 nm for Trp,49 ∼35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 cm−1 M−1 at 280 for HSA,49 38940 cm−1 M−1 at 280 nm for lysozyme,50 6200 cm−1 M−1 at 276 nm for insulin,51 and 1295 cm−1 M−1 at 278 nm for Tyr.49
200 cm−1 M−1 at 280 for HSA,49 38940 cm−1 M−1 at 280 nm for lysozyme,50 6200 cm−1 M−1 at 276 nm for insulin,51 and 1295 cm−1 M−1 at 278 nm for Tyr.49
Poly- and oligo-peptide concentrations were determined by weighing the dissolved powder in the case of octa-L-arginine (DK), by absorbance at 214 nm for Poly-K (IT), using an extinction coefficient of 923 cm−1 M−1 for the monomer,52 or by the bicinchoninic acid method53 for Poly-A and Poly-K solutions (MY), using bovine serum albumin as a standard, whose concentration was determined spectrophotometrically, with an extinction coefficient of ∼43900 cm−1 M−1 at 280 nm.49
Concentrations used in the 3D spectra were: HSA 0.9 μM (IT). 1.5 μM (DK) or 3 μM (MY); lysozyme 4 μM (MY); insulin 17 μM (DK); Trp 0.9 μM (IT) or 3 μM (MY); Tyr 3 μM; Poly-A 0.2 mg ml−1; octa-L-arginine 0.2 mg ml−1; Poly-K 0.2 mg ml−1 (including the Br− counter ion).
Fluorescence spectroscopy
Fluorescence spectra were collected with a Cary Eclipse fluorescence spectrometer (Agilent Technologies) (IT and DK), or a FP-6500 spectrofluorimeter (Jasco) (MY). Both these instruments have also been used by several of the authors claiming the presence of backbone fluorescence. Experiments were performed in 1 cm path length cells. 3D fluorescence spectra were executed under the following conditions: excitation 220–350 nm; emission 220–450 nm; data interval 2 nm (IT, DK) or 5 nm in excitation and 1 nm in emission (MY); bandwidth 2.5 nm (IT), 5 nm (DK) or 10 nm (MY); scanning rate 600 nm min−1 (IT), 1200 nm min−1 (DK) or 500 nm min−1 (MY). In the Cary the detector voltage was set at 800 V (IT) or 600 V (DK), while it was fixed at 250 V (MY) in FP-6500. Blank subtraction or spectral corrections were not performed. Fluorescence emission or excitation spectra were executed under the same conditions of the 3D spectra (DK) or with the following settings (IT): data interval 1 nm, excitation bandwidth 5 nm, emission bandwidth 10 nm, scan rate 30 nm min−1, detector voltage 600 V. Excitation spectra were corrected for inner filter effects.54Results and discussion
3D fluorescence spectra of proteins
Fig. 1 reports the normalised 3D fluorescence spectra/contour plots of three representative proteins: HSA, lysozyme and human insulin. These molecules are very diverse in size (585, 129 and 51 amino acid residues, respectively), and aromatic amino acid composition: Phe dominates in HSA (1 Trp, 18 Tyr and 31 Phe), Trp in lysozyme (6 Trp, 3 Tyr and 3 Phe), while insulin only contains Tyr and Phe (4 Tyr and 2 Phe).The 3D fluorescence spectra of HSA collected in the three laboratories participating in the present study show that the relative intensities of the different peaks and even their exact position can vary due to inner filter effect and instrumental factors, such as the wavelength response of monochromators and detectors. Corrections for these factors are in principle possible,1,55 but were not performed in the current study, as it is customary to report uncorrected, so-called “technical” spectra, particularly for 3D fluorescence.1 Therefore, the multiple datasets reported here serve to illustrate possible variations in the spectral shape, due to instrumental factors. Indeed, a variability in exact peak positions and relative intensities similar to those observed in our three labs can also be noticed among the papers claiming backbone fluorescence.10–26
Some of the signals in the 3D fluorescence spectra are not due to fluorescence, but to light scattering, and can therefore be observed also for a sample that does not contain any fluorophore, such as buffer or pure water. The line labelled as ‘a’, corresponding to the matrix diagonal, i.e. the region with λem. = λexc., is due to elastic scattering of the excitation light by the sample solvent, the dissolved molecules, and suspended particulates. A second line, labeled as ‘b’, is observed at λem. = 2λexc., and is due to an instrumental artefact caused by transmission of second-order diffraction by grating monochromators. That is, when set to select a given wavelength, λ, a grating monochromator also partially transmits light with wavelength λ/2.1,56 For these reasons, peak ‘b’ actually corresponds to light of wavelength λem. = λexc. that can cross the emission monochromator, when this is set at λem. = 2λexc..
Another much weaker line is sometimes visible (depending on the fluorescence intensity scale), and is due to inelastic (Raman) scattering by the solvent. In this phenomenon, photons lose an amount of energy corresponding to the energy needed to excite vibrations of the solvent molecules. Therefore, the energy difference between the incident and scattered light (as the wavelength of the first is varied in a scan) is constant. A constant energy difference corresponds to a varying difference in wavelengths. Therefore, Raman scattering is responsible for the line labelled as ‘c’ in the Fig. 1, which runs close to line ‘a’, but is not parallel to it.
The remaining two peaks, designated as peaks ‘1’ and ‘2’ in the 3D fluorescence spectra, are due to protein fluorescence. The comparison of the three HSA 3D fluorescence spectra shows that the relative intensities of these two peaks with respect to the scattering signals is dependent on protein concentration (in addition to the aforementioned experimental factors).
Peak ‘1’ corresponds to excitation at ∼280 nm and emission at 330–340 nm (for HSA and lysozyme) or 304 nm for insulin. This peak corresponds to emission by the aromatic side-chains of Trp and Tyr residues in proteins. Both these amino acids have an absorbance peak at ∼280 nm, and are known to be significantly fluorescent, while emission from Phe can be neglected.30 Tyr fluorescence is insensitive to the polarity of its environment, and its emission maximum is always found around 303–305 nm.1,2,57 Indeed, insulin contains four Tyr residues, and no Trp, and in its 3D spectrum peak ‘1’ is located at λem. = 304 nm. By contrast, Trp emission is very sensitive to the polarity of its environment and its fluorescence peak shifts from above 350 nm in water down to almost 305 nm in very nonpolar environments or when buried in a hydrophobic protein core,1,2 like in the extreme case of azurin.58 The quantum yields of Tyr and Trp fluorescence are comparable. However, Tyr fluorescence in proteins is usually rather weak, mostly for two reasons: its extinction coefficient at 280 nm is lower by a factor of four than that of Trp, and its emission partially overlaps with the absorption spectrum of Trp, so that Förster resonance energy transfer (FRET) can occur from Tyr to Trp residues. Consistent with these considerations, peak ‘1’ in HSA and lysozyme is dominated by Trp emission, and is located at λem. values, typical for this amino acid, with a red shifted maximum for lysozyme, where the Trp residues are more solvent exposed than the lone Trp residue of HSA.
Peak ‘2’ is the feature of protein 3D fluorescence spectra that has been claimed to be due to protein backbone emission. By comparing the 3D fluorescence spectra of Fig. 1, we note that the wavelength of maximum emission for this peak is always essentially coincident with that of peak ‘1’, even though the latter varies significantly from one protein to the other (HSA MY: peak ‘1’, 330 nm; peak ‘2’, 331 nm; HSA DK: peak ‘1’, 336 nm; peak ‘2’, 340 nm; HSA IT: peak ‘1’, 332 nm; peak ‘2’, 334 nm; insulin: peak ‘1’, 304 nm, peak ‘2’, 302 nm; lysozyme: peak ‘1’, 339 nm, peak ‘2’, 342 nm). This spectral similarity of the fluorescence is further exemplified by overlaying the emission spectra of HSA and insulin upon excitation at 220 and 280 nm in Fig. 2. Kasha's rule1,2,59 states that the position and shape of the emission spectrum is independent of the excitation wavelength, since fluorescence occurs in appreciable yield only from the lowest excited singlet state. Therefore, the observation of a coincidence of the maximum emission wavelength for peaks ‘1’ and ‘2’ is the first indication that the fluorescence originates from the excitation of the same fluorophore at different excitation wavelengths, and immediately suggests that peak ‘2’ is due to excitation of a higher excited state of the aromatic residues.
 | ||
| Fig. 2 Normalised emission spectra (DK) of: (left panel) the proteins HSA (red) and insulin (blue), and (right panel) the aromatic amino acids Trp (dark red) and Tyr (light blue), excited at 220 (dashed lines) and 280 nm (solid lines). | ||
Peak ‘2’ is due to aromatic residues
To further show that peak ‘2’ is due to the aromatic moieties and not the protein backbone, we measured the 3D fluorescence spectra of individual aromatic amino acids, which therefore contain no amide bonds. Fig. 3 shows that peak ‘2’ is present also for solutions of Trp and Tyr and that its position corresponds roughly to those observed for Trp containing proteins and for proteins with Tyr residues only, respectively (Trp 353 nm; Tyr 303 nm).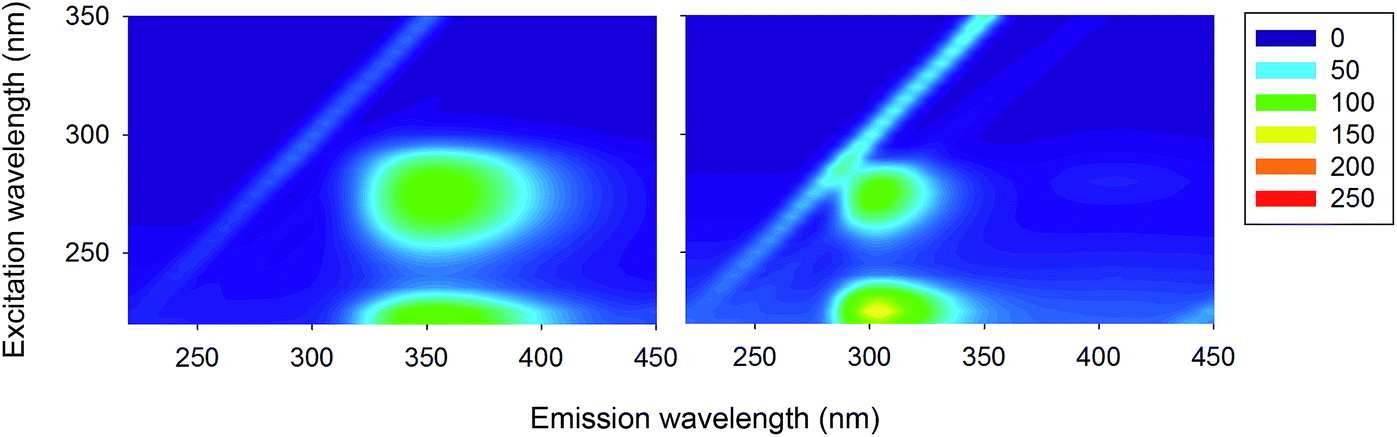 | ||
| Fig. 3 Normalised 3D fluorescence spectra (as contour maps) of Trp (left) and Tyr (right) solutions (MY). The spectra were normalised to 100 on peak ‘1’. | ||
There is a significant difference for the observed emission maximum of lysozyme and HSA (∼330–340 nm) and that of Trp (∼350 nm), but this can be explained by the different solvent exposure of Trp in the various samples.1,2,31
Also in the case of Trp and Tyr, overlap of the normalised emission spectra obtained with excitation at 220 nm and 280 nm (Fig. 2) confirms that the emitting species is the same. Finally, Fig. 4 reports the excitation and absorption spectra for HSA and Trp, collected by measuring the emission intensity at 350 nm. For both samples, two bands are observed, peaked at approximately 280 and 220 nm, confirming that the 3D peak observed in the case of HSA and often attributed to backbone emission, actually originates from the fluorescence of aromatic residues.30 The slight differences between the excitation spectra of Trp and HSA can be attributed to the presence of Tyr residues in the protein and to the occurrence of Tyr-to-Trp energy-transfer, which contributes to the observed fluorescence.
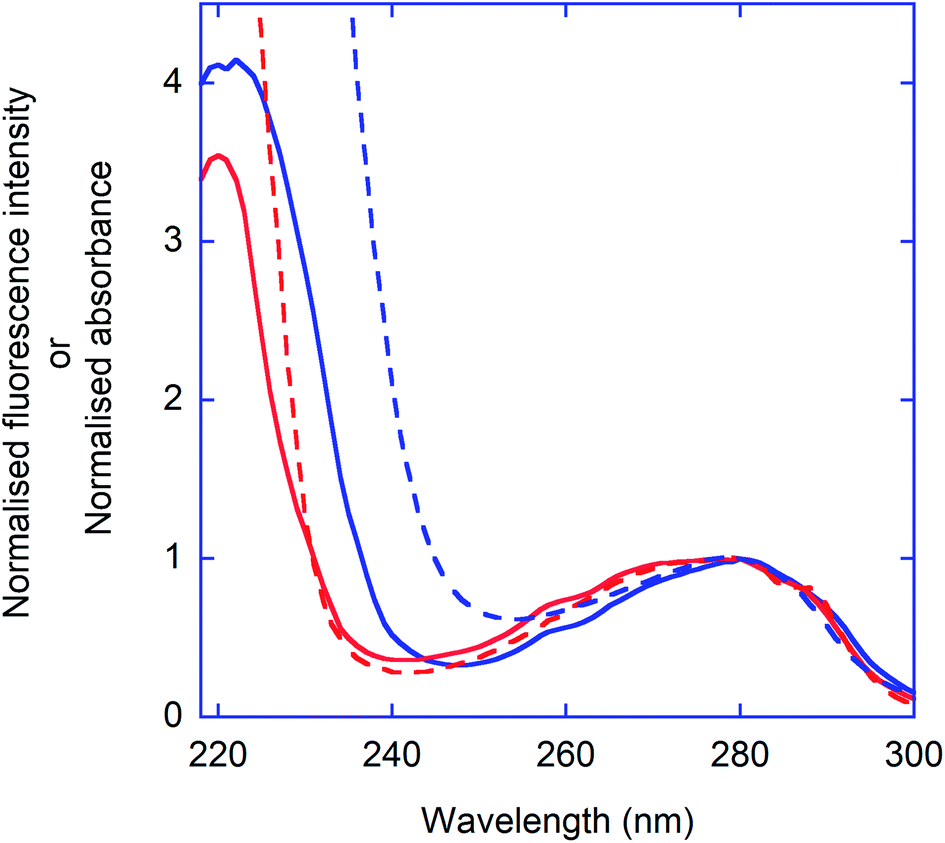 | ||
| Fig. 4 Excitation spectra (IT) of Trp (red solid line) and HSA (blue solid line). For comparison, the absorption spectrum of Trp and HSA are also reported (dashed lines). λem. = 350 nm. The spectra were normalised to 1 at 280 nm. | ||
The identity of the excited states corresponding to the two excitation peaks of Trp and Tyr (and therefore also of proteins) has been the object of extensive experimental and theoretical studies. Near UV light (∼280 nm) leads to excitation of quasi-degenerate excited states termed 1La and 1Lb in Platt's notation. Excitation in the far UV (peak at 220 nm) brings molecules in higher energy states called 1Ba and 1Bb.60–62 A small fraction of these molecules then can undergo photoionization, but most relax very rapidly to 1La and 1Lb, with subsequent fluorescence emission.63,64
The protein backbone is non-fluorescent
As a final confirmation that the peptide backbone does not contribute at all to protein fluorescence, we measured 3D fluorescence spectra for a number of homopolypeptides containing no aromatic residues (Fig. 5). In these cases, the 3D fluorescence spectra correspond to the spectrum observed for the buffer alone (Fig. 1, bottom row, right), and contain only scattering signals. That is, no fluorescence signal is observed in the region 300–350 nm, as is observed for the proteins containing aromatic residues.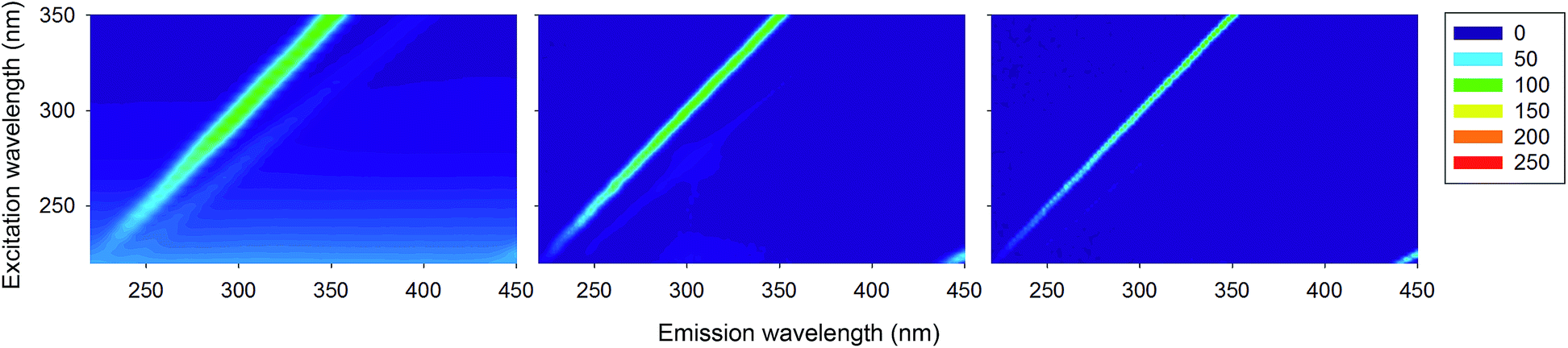 | ||
| Fig. 5 Normalised 3D fluorescence spectra (as contour maps) of poly-L-alanine (MY, left), oligo-L-Arg (DK, center) and poly-L-Lys (IT, right), normalised to 100 on the maximum of the ‘a’ band. | ||
Conclusions
Even though the polypeptide backbone absorbs in the far-UV region, we show here that it emits no fluorescence. The emission peak at 300–350 nm, observed in 3D fluorescence spectra upon excitation at 220–230 nm, often claimed to be caused by backbone emission, is in reality due to the excitation of higher excited electronic states of the aromatic residues present in the protein. Once the aromatic moieties reach these states by absorption of far-UV light, they rapidly relax to the lowest excited state and from there emit at the usual wavelengths of 300–350 nm, according to Kasha's rule. Therefore, this peak reports simply on the local environment of the aromatic residues, just like the peak with excitation at 280 nm, and not on the backbone conformation or secondary structure of the protein.As illustrated in the introduction, this conclusion should have been obvious to any expert in protein fluorescence. Nonetheless, this error has persisted for more than 8 years, and we estimate that there are now well over 100 papers claiming to have observed fluorescence from the protein backbone. Unfortunately, we are witnessing an alarming number of errors or misinterpretations in several published fluorescence studies (for a discussion of this issue, see ref. 9, 54, 56 and 65–67). The current paper hopefully contributes to a reassessment of one of the possible pitfalls.
Acknowledgements
ST thanks the University of Malaya (University Malaya Research Grant, Grant Number: RG275-14AFR) for supporting this research.References
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 Search PubMed.
- D. M. Jameson, Introduction to Fluorescence, CRC Press, New York, 2014 Search PubMed.
- D. W. Johnson, J. B. Callis and G. D. Christian, Anal. Chem., 1977, 49, 747–757 CrossRef.
- J. H. Rho and J. L. Stuart, Anal. Chem., 1978, 50, 620–625 CrossRef CAS.
- E. R. Weiner, Anal. Chem., 1978, 50, 1583–1584 CrossRef CAS.
- F. F. Hartline, Science, 1979, 4387, 1330–1331 Search PubMed.
- Á. Andrade-Eiroa, M. Canle and V. Cerdá, Appl. Spectrosc. Rev., 2013, 48, 1–49 CrossRef.
- E. M. Carstea, J. Bridgeman, A. Baker and D. M. Reynolds, Water Res., 2016, 95, 205–219 CrossRef CAS PubMed.
- L. Stella, M. van de Weert, H. D. Burrows and R. Fausto, J. Mol. Struct., 2014, 1077, 1–3 CrossRef CAS.
- S. Huang, S. Peng, W. Su, Z. Tang, J. Cui, C. Huang and Q. Xiao, RSC Adv., 2016, 6, 47043–47054 RSC.
- F. Ahmad, Y. Zhou, Z. Ling, Q. Xiang and X. Zhou, RSC Adv., 2016, 6, 35719–35730 RSC.
- T. S. Banipal, A. Kaur, I. A. Khan and P. K. Banipal, RSC Adv., 2016, 6, 34754–34769 RSC.
- S. Huang, H. Qiu, J. Xie, C. Huang, W. Su, B. Hu and Q. Xiao, RSC Adv., 2016, 6, 44531–44542 RSC.
- J. He, H. Yang, S. Li, K. Xu, Q. Wang, Y. Huang and H. Li, RSC Adv., 2016, 6, 61119–61128 RSC.
- A. M. Alanazi and A. S. Abdelhameed, PLoS One, 2016, 11, e0146297 Search PubMed.
- S. Chatterjee and G. S. Kumar, J. Photochem. Photobiol., B, 2016, 159, 169–178 CrossRef CAS PubMed.
- L. Jattinagoudar, M. Meti, S. Nandibewoor and S. Chimatadar, Spectrochim. Acta, Part A, 2016, 156, 164–171 CrossRef CAS PubMed.
- Y. Li, Q. Wang, J. He, J. Yan and H. Li, Luminescence, 2016, 31, 38–46 CrossRef CAS PubMed.
- T. Li, Z. Cheng, L. Cao and X. Jiang, Food Chem., 2016, 194, 740–748 CrossRef CAS PubMed.
- K. Mitra, S. Singh, S. K. Hira, V. K. Patel, D. Singh, S. Vishwakarma, R. Singh, A. Kumari, P. P. Manna and B. Ray, ACS Biomater. Sci. Eng., 2016, 2, 1630–1640 CrossRef CAS.
- A. S. Roy and P. J. Ghosh, J. Inclusion Phenom. Macrocyclic Chem., 2016, 84, 21–34 CrossRef.
- A. Salci and M. Toprak, J. Biomol. Struct. Dyn., 2016 DOI:10.1080/07391102.2015.1128357.
- L.-H. Wang, M.-S. Wang, X.-A. Zeng, D. Gong and Y.-B. Huang, Biochim. Biophys. Acta, 2016 DOI:10.1016/j.bbagen.2016.08.002.
- J. Yan, B. Tang, D. Wu, S. Li, K. Xu, X. Ma, Q. Wang and H. Li, Spectrosc. Lett., 2016, 49, 541–549 CrossRef.
- H. Yang, Y. Huang, D. Wu, J. Yan, J. He and H. Li, New J. Chem., 2016, 40, 2530–2540 RSC.
- R. Zhang, R. Liu and W. Zong, J. Agric. Food Chem., 2016, 64, 6630–6640 CrossRef CAS PubMed.
- D. Pinotsi, L. Grisanti, P. Mahou, R. Gebauer, C. F. Kaminski, A. Hassanali and G. S. Kaminski Schierle, J. Am. Chem. Soc., 2016, 138, 3046–3057 CrossRef CAS PubMed.
- P. Debye and J. O. Edwards, Science, 1952, 116, 143–144 CAS.
- G. Weber, Adv. Protein Chem., 1953, 8, 415–459 CrossRef CAS PubMed.
- F. W. Teale and G. Weber, Biochem. J., 1957, 65, 476–482 CrossRef CAS PubMed.
- F. W. Teale, Biochem. J., 1960, 76, 381–388 CrossRef CAS PubMed.
- S. V. Konev, Fluorescence and Phosphorescence of Proteins and Nucleic Acids, Plenum Press, New York, 1967 Search PubMed.
- C. Goletz, M. Wagner, A. Grübel, W. Schmidt, N. Korf and P. Werner, Talanta, 2011, 85, 650–656 CrossRef CAS PubMed.
- C. Bonnin, M. Matoga, N. Garnier, C. Debroche, B. de Vandière and P. Chaminade, J. Chromatogr., 2007, 1156, 94–100 CrossRef CAS PubMed.
- S. Determann, J. M. Lobbes, R. Reuter and J. Rullkeotter, Mar. Chem., 1998, 62, 137–156 CrossRef CAS.
- Y. Z. Zhang, B. Zhou, Y. X. Liu, C. X. Zhou, X. L. Ding and Y. Liu, J. Fluoresc., 2008, 18, 109–118 CrossRef CAS PubMed.
- A. Credi and L. Prodi, J. Mol. Struct., 2014, 1077, 30–39 CrossRef CAS.
- S. R. Feroz, S. B. Mohamad, Z. S. D. Bakri, S. N. A. Malek and S. Tayyab, PLoS One, 2013, 8, e76067 CAS.
- J. R. Albert-Garcia, G. M. Antón-Fos, M. J. Duart, L. Lahuerta Zamora and J. Martínez Calatayud, Talanta, 2009, 79, 412–418 CrossRef CAS PubMed.
- B. Valeur and M. N. Berberan-Santos, Molecular Fluorescence, Wiley VCH, New York, 2012 Search PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, California, 2010 Search PubMed.
- B. Wardle, Principles and Applications of Photochemistry, Wiley VCH, New York, 2009 Search PubMed.
- D. B. Wetlaufer, Adv. Protein Chem., 1963, 17, 303–390 CrossRef.
- K. Rosenheck and P. Doty, Proc. Natl. Acad. Sci. U. S. A., 1961, 47, 1775–1785 CrossRef CAS.
- G. G. Stokes, Philos. Trans. R. Soc. London, 1858, 142, 463–562 CrossRef.
- C. Reichardt and T. Welton, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, 4th edn, 2010 Search PubMed.
- L. Cwiklik, A. J. A. Aquino, M. Vazdar, P. Jurkiewicz, J. Pittner, M. Hof and H. Lischka, J. Phys. Chem. A, 2011, 115, 11428–11437 CrossRef CAS PubMed.
- D. Petsalakis, D. G. Georgiadou, M. Vasilopoulou, G. Pistolis, D. Dimotikali, P. Argitis and G. Theodorakopoulos, J. Phys. Chem. A, 2010, 114, 5580–5587 CrossRef PubMed.
- C. N. Pace, F. Vajdos, L. Fee, G. Grimsley and T. Gray, Protein Sci., 1995, 4, 2411–2423 CrossRef CAS PubMed.
- R. C. Davies, A. Neuberger and B. M. Wilson, Biochim. Biophys. Acta, 1969, 178, 294–305 CrossRef CAS.
- J. Brange, U. Ribel, J. F. Hansen, G. Dodson, M. T. Hansen, S. Havelund, S. G. Melberg, F. Norris, K. Norris, L. Snel, A. R. Sørensen and H. O. Voigt, Nature, 1988, 333, 679–682 CrossRef CAS PubMed.
- B. J. H. Kuipers and H. Gruppen, J. Agric. Food Chem., 2007, 55, 5445–5451 CrossRef CAS PubMed.
- R. E. Brown, K. L. Jarvis and K. J. Hyland, Anal. Biochem., 1989, 180, 136–139 CrossRef CAS PubMed.
- M. van de Weert and L. Stella, J. Mol. Struct., 2011, 998, 144–150 CrossRef CAS.
- J. W. Hofstraat and M. J. Latuhihin, Appl. Spectrosc., 1994, 48, 436–447 CrossRef CAS.
- L. Stella, A. L. Capodilupo and M. Bietti, Chem. Commun., 2008, 39, 4744–4746 RSC.
- J. B. A. Ross, W. R. Laws, K. W. Rousslang and H. R. Wyssbrod, in Topics in Fluorescence Spectroscopy: Biochemical Applications, ed. J. R. Lakowicz, Plenum Press, New York, 1992, vol. 3, ch. 1, pp. 1–63 Search PubMed.
- G. Gilardi, G. Mei, N. Rosato, G. W. Canters and A. Finazzi-Agrò, Biochemistry, 1994, 33, 1425–1432 CrossRef CAS PubMed.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- B. Albinsson and B. Nordén, J. Phys. Chem., 1992, 96, 6204–6212 CrossRef CAS.
- A. C. Borin and L. Serrano-Andrés, Chem. Phys., 2000, 262, 253–265 CrossRef CAS.
- D. M. Rogers and J. D. Hirst, J. Phys. Chem. A, 2003, 107, 11191–11200 CrossRef CAS.
- H. B. Steen, J. Chem. Phys., 1974, 61, 3997–4002 CrossRef CAS.
- G. Köhler and N. Getoff, Chem. Phys. Lett., 1974, 26, 525–528 CrossRef.
- M. Van De Weert, J. Fluoresc., 2010, 20, 625–629 CrossRef CAS PubMed.
- S. Bobone, M. van de Weert and L. Stella, J. Mol. Struct., 2014, 1077, 68–76 CrossRef CAS.
- A. A. Sousa, J. Fluoresc., 2015, 25, 1567–1575 CrossRef CAS PubMed.
Footnotes |
| † These authors contributed equally to this work. |
| ‡ Current address: Novo Nordisk A/S, Novo Nordisk Parken 1, 2760 Måløv, Denmark. |
| § Current address: Humboldt Universität zu Berlin, Institut für Biologie, 10099 Berlin, Germany. |
| This journal is © The Royal Society of Chemistry 2016 |