
A universal surface enhanced Raman spectroscopy (SERS)-active graphene cathode for lithium–air batteries†
Kewei Liu‡
,
Zitian Yu‡,
Xiaowen Zhu,
Shuo Zhang,
Feng Zou and
Yu Zhu*
Department of Polymer Science, The University of Akron, 170 University Circle, Akron, Ohio 44325-3909, USA. E-mail: yu.zhu@uakron.edu
First published on 7th October 2016
Abstract
Nonaqueous lithium–air (Li–O2) batteries, with their high theoretical energy densities far exceeding those of conventional Li-ion batteries, have attracted significant research interest over the past decade. However, the practical realization of Li–O2 batteries is still confronted with the challenge of electrode side reactions that lead to severe solvent/electrode degradation upon cycling. To understand the reaction process on the electrode, it is necessary to obtain detailed information about the chemicals formed on the Li–O2 battery electrode. Herein, a universal method to fabricate large-area regularly patterned gold nano-dots using an anodic aluminum oxide (AAO) mask was developed. The gold nano-dots were patterned on to conductive substrates such as Au film and graphene, and then the films were used as SERS (Surface Enhanced Raman Spectroscopy)-active cathodes in Li–O2 batteries. The discharge products on the different electrodes (graphene and gold) were analyzed and the results indicated that the SERS electrode will be a useful tool for studying the reaction process on lithium–air cathodes.
Introduction
Among all the advanced electricity energy storage systems, metal–air batteries, especially lithium–air batteries, comprise a new system that has the potential to significantly enhance the energy density. It has been shown by theoretical work that their energy densities are comparable to that of gasoline.1,2 Experimentally, a rechargeable battery with a capacity of 2000 mA h g−1 (100 cycles) has also been reported.3 However, despite the rapid advances during the past few years, current lithium–air batteries are still an underdeveloped system for use in practical applications.1,4–13 The biggest challenge is that lithium–air batteries undergo severe degradation upon cycling. A wealth of research has suggested that intermediate compounds, especially the superoxide anion radicals,9 react with solvent molecules and generate irreversible byproducts that eventually destroy the cycling ability of the battery. More recently, several studies have also elucidated that the carbon based electrode, the most explored cathode for lithium–air batteries, may promote reactions with the superoxide species14–16 and result in irreversible charge/discharge processes even with improved electrolyte systems. As such, the investigation of the electrochemical process during charging and discharging cycles becomes important. Because the reactions on the lithium–air batteries’ cathode are very complicated, characterization of the electrode products is generally difficult. X-ray diffraction (XRD)16 and Raman spectroscopy16–18 are the most common techniques for identifying the discharge products such as lithium peroxide and lithium carbonate.19 However, those techniques have limited capabilities to detect some side-reactions, for instance as reported previously,19 XRD shows that the only discharge product is lithium peroxide, yet other techniques (NMR and FTIR) indicate that side-reaction products such as lithium carbonates existed. Raman spectroscopy is a useful tool to investigate electrode products in batteries. However, as pointed out by some researchers,9 the major challenge of Raman spectroscopy is weak Raman signals, which result in poor resolution of the Raman spectra obtained from the cathode directly. In this scenario, a sensitive and convenient method to investigate the lithium oxygen battery electrode reaction is highly desired.Surface enhanced Raman spectroscopy (SERS)20–23 is an effective method to detect trace intermediate and product species on the electrode. SERS utilizes the effect by which Raman signals on certain metal surfaces can be dramatically enhanced due to the enhancement of the electrical field (electromagnetic enhancement) and/or the enhancement of polarizability (chemical enhancement).20,22,23 Such metal surfaces are usually small silver or gold colloids, electrochemically-roughened silver/gold electrodes etc. Because gold is a stable material in lithium–air batteries,16 it is well suited to modifying the lithium–air battery cathode to get enhanced Raman signals. However, traditional Au SERS-active electrodes were created by casting Au nano-particles on an electrode surface,24 sputtering Au as an electrode25 or electrochemically roughening Au surfaces,18,26–28 during which random “hot spots” were formed to enhance the Raman signals.29–31 Therefore, a SERS electrode with a uniform SERS effect over the entire electrode was desired to explore the reaction mechanism on the electrode. In this work, a SERS-active electrode with highly ordered gold nanoparticles was prepared and used as a lithium–air battery cathode. The results indicated that such a SERS-active electrode enhances the Raman signals over the entire electrode, rendering it a powerful tool to investigate all species formed on the cathode. This SERS-active electrode is used to further understand the reaction process in lithium–air batteries.
Experimental section
Preparation of the AAO masks
A high-purity aluminum (Al) sheet (thickness 0.25 mm, purity 99.997%, Alfa Aesar) was cut into rectangular samples (10 × 20 mm). The Al sheet was degreased in acetone with ultrasonic agitation (Ultrasonic cleaner GB-2500B, Bringnew) for 3 min then rinsed with DI water. After that, the Al sheet was chemically polished in 5% wt NaOH solution at 60 °C for 30 s then rinsed with DI water. The clean Al sheet was electrochemically polished in a mixed solution32 (14.9 mL DI water + 70 mL ethanol + 10 mL 2-butoxyethanol + 15.3 mL perchloric acid) under 500 mA at 0 °C for 90 s. The sample was then rinsed with DI water. Two-step anodization was used to achieve highly ordered AAO templates.32 For the AAO mask with small pores, the polished Al sheet was anodized in 3% wt oxalic acid under 40 V at 0 °C for 7.5 min. The sample was then immersed in a mixed solution of chromic acid and phosphoric acid (0.2 M H2CrO4 and 0.4 M H3PO4) at 60 °C for 5 min to remove the oxidized layer. The second anodization was also carried out in 3% wt oxalic acid at 0 °C for 6 min to form parallel nanopores on the surface. For the AAO mask with large pores, the polished Al sheet was anodized in 10% wt phosphoric acid under 140 V at 0 °C for 6 min. The sample was then immersed in a mixed solution of chromic acid and phosphoric acid at 60 °C for 6 min to remove the oxidized layer. The second anodization was conducted in phosphoric acid at 140 V and 0 °C again for 6 min to prepare the AAO mask with nanochannels. After anodization, a layer of poly(methyl methacrylate) (PMMA) solution (4% wt PMMA in anisole, Mw = 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was spin-cast onto the AAO surface at 1000 rpm. Then the Al was removed by floating the sample on 3% wt CuCl2 solution. In order to open and widen the pores, the AAO mask was washed with a mixed solution of chromic acid and phosphoric acid (0.2 M H2CrO4 and 0.4 M H3PO4) at 60 °C. After rinsing with deionized water, the AAO masks were transferred onto different substrates (silicon wafer, gold and graphene).
000 g mol−1) was spin-cast onto the AAO surface at 1000 rpm. Then the Al was removed by floating the sample on 3% wt CuCl2 solution. In order to open and widen the pores, the AAO mask was washed with a mixed solution of chromic acid and phosphoric acid (0.2 M H2CrO4 and 0.4 M H3PO4) at 60 °C. After rinsing with deionized water, the AAO masks were transferred onto different substrates (silicon wafer, gold and graphene).
Preparation of SERS-active substrates
Three SERS-active substrates (silicon wafer, gold, graphene) were prepared in this work. Silicon wafers (4 inch P-type, University Wafer) were cleaned by sonicating in acetone for 10 min followed by ethanol and DI water or another 10 min. Gold substrates were prepared by sputtering gold (∼50 nm) onto Si wafers. Graphene was grown by a chemical vapor deposition33 method and then transferred onto the gold substrate. After the AAO masks were transferred to the substrates, the PMMA on the AAO masks was removed by acetone washing. Then the samples were dried, and 20 nm thick gold was evaporated (evaporation rate 1 Å s−1) onto the substrates through the AAO masks by an e-beam evaporator. After gold deposition, the AAO membranes were removed by 0.1 M (NH4)2S2O8 at 60 °C. Rhodamine 6G was dissolved in ethanol to prepare solutions with concentrations of 10−3 M and 10−6 M. Fresh R6G solution was drop-cast on to the SERS substrate and then used for Raman spectroscopy characterization. A HORIBA LabRAM HR Raman microscope equipped with a frequency doubled Nd:YAG laser (532 nm/25 mW) was used to collect the Raman spectra. The acquisition time was 10 s and the scans were cycled 3 times. For the Raman mapping experiment, the same configuration of the laser was used. The mapping step was 2 μm in an area of 20 × 20 μm. Raman spectroscopic analysis of the lithium–air battery electrode was conducted as follows: the discharged working electrode was transferred into a glovebox and rinsed with DMSO to remove residual salts. After the electrode was dried, it was sealed by cover slips with epoxy glue inside the glovebox. The sealed sample was used for Raman spectroscopic characterization so that the ambient environment would not change the components formed on the electrode surface.Fabrication of lithium–air batteries
A three-electrode system was used for the lithium–air battery test. The SERS electrodes were used as the working electrode. Lithium metal attached onto a stainless steel mesh served as the reference electrode. Carbon cloth (Fuel Cell Earth, CCP10) was used as the counter electrode. 1 M LiTFSI (99.6%, Matrix) in DMSO (99.8% EMD) was used as the electrolyte. Pure oxygen (Praxair) was purged into the system for 30 min before the electrochemical test.SEM/AFM/TEM characterization
Samples were directly imaged by a JEOL-7401 Field Emission Scanning Electron Microscope and Atomic Force Microscopy. The height images were recorded on a Digital Instruments MultiMode Scanning Probe Microscopy (SPM) system under tapping mode. For TEM characterization, gold nano-dots on freestanding graphene were prepared as follows: the AAO mask was transferred onto a monolayer of graphene grown on Cu foil, after gold evaporation, the gold nano-dots on the graphene were obtained by removal of AAO by 5% wt NaOH at 60 °C. Cu, as the graphene substrate, was etched in 0.1 M (NH4)2S2O8. Then the graphene film with patterned gold nano-dots was floated on water to remove the salts. A piece of the graphene patterned with gold nano-dots can be collected by using a TEM grid. The sample was dried under vacuum for TEM characterization. A JEOL 1203 Scanning Transmission Electron Microscope was used to collect all the TEM images in this work.Results and discussion
In order to fabricate a SERS-active electrode with highly ordered gold nanoparticles, a universal method of metal deposition through a nano-shadow mask was developed in this work. The nano-shadow mask needs to have a highly ordered structure and small features to obtain a good SERS effect. Thus, anodic aluminum oxide (AAO) masks34 were chosen. AAO can form a highly ordered structure and it has been reported to be able to produce silver nano-voids,35 aligned silver nano-rods,36 silver nano-dot arrays37 and gold nano-dot arrays.34,38,39Scheme 1 shows the preparation procedure of the SERS-active electrodes. AAO is prepared by anodization of aluminum in solution.32 The anodized alumina can be removed from the aluminum substrate by treatment with a CuCl2 solution.40 However, the anodized alumina nanotubes are closed-ended at the bottom. In order to use them as a mask, the ends must be opened. This is done by additional etching with chromic acid.34 After the free-standing AAO mask is prepared, the gold dots are fabricated on the substrate by metal deposition through the AAO mask.
 | ||
| Scheme 1 Schematic illustration of the gold nano-dot array preparation on different surfaces. The AAO template was prepared by polishing and anodization of aluminum foil. After the bottom of the AAO template was etched, the AAO mask was transferred on to arbitrary substrates (Si wafer, graphene or gold) and gold nano-dot arrays were deposited. The mask was then removed to form gold nano-dot modified substrates. The Si wafer substrate with a gold nano-dot array was used for the standard SERS analysis. The gold and graphene substrates with gold nano-dot arrays were used as SERS-active electrodes in the Li–O2 battery study. | ||
The size of the gold dots is important for the enhancement of Raman spectroscopy. Therefore, AAO masks with different pore sizes were prepared in this work by controlling the anodization voltage and the acidic media.32 With oxalic acid and an anodization voltage of 40 V, an AAO membrane with an average pore size of 80 nm was obtained. When the acid was switched to phosphoric acid and the anodization voltage was increased to 120 V, an AAO membrane with an average pore size of 300 nm was formed. After anodization, CuCl2 was used to remove the aluminum and chromic acid was used to open the bottom. The free standing AAO masks are shown in Fig. S1,ESI.†
The free-standing AAO masks were transferred onto the target substrate and gold was deposited on to the substrate through the mask to form regularly arranged gold dots. The AAO mask was removed by an ammonia persulfate ((NH4)2S2O8) etching solution. As the purpose of this work is to fabricate SERS-active electrodes for lithium–air battery studies, several substrates were explored in this work. Bare silicon wafers were first used to examine the morphology of the gold dots, as well as serve as the standard substrate for SERS analysis. Then the conductive substrates (gold and graphene) were used for SERS analysis of the battery electrode. Gold and graphene were selected in this work as they represent two of the most investigated cathode materials (roughened gold and various carbon electrodes)16,41 in lithium–air battery research.
The morphology of the gold nano-dots on silicon wafers, gold and graphene substrates is shown in Fig. 1. Fig. 1a–c are SEM images of the gold dots fabricated with AAO masks with small pores. The average diameter of the gold dots is about 80 nm. The results indicated that the gold nano-dots are uniformly distributed on the surface regardless of the properties of the substrates. Fig. 1d–f demonstrate the large gold nano-dots prepared with the AAO mask with large pores. The average diameter of the large gold nano-dots is around 300 nm. In principle, this AAO mask based method can be used to prepare regularly assembled metal nano-dots on any arbitrary substrate. The size of the samples easily reaches the centimeter scale, which allows them to be used in battery devices.
 | ||
| Fig. 1 SEM images of the gold nano-dots obtained from AAO templates on different substrates (the inset images are the same samples under higher magnification). (a–c) Small gold nano-dots on different substrates as noted. (d–f) Large gold nano-dots on different substrates as noted. The scale bar in (a–f) is 1 μm. The scale bar of the inset images for the small gold nano-dots and the large gold nano-dots is 200 nm and 500 nm, respectively. | ||
The gold nano-dots on different substrates were also examined by atomic force microscopy (AFM). As shown in Fig. 2a–c, small gold nano-dots prepared on silicon wafer, gold and graphene substrates have a height of ∼20 nm (ESI Fig. S2†), which is consistent with the thickness of the gold deposition. A similar thickness was observed for the gold nano-dots prepared by the AAO mask with larger pore sizes (ESI Fig. S3†). For the samples of gold nano-dots on graphene, the graphene was grown on copper by a chemical vapor deposition method,31 then the graphene film was transferred onto the silicon wafer surface for gold nano-dot deposition. The three-dimensional AFM image of the gold nano-dots on a graphene sample is shown in Fig. 2d. Freestanding gold nano-dots on graphene samples were prepared as well. These allow the sample to be characterized by transmission electron microscopy (TEM). The TEM images of the large gold nano-dots (Fig. 2e) and the small gold nano-dots (Fig. 2f) on graphene clearly confirmed that such a SERS-active electrode can be prepared by a facile deposition method. As far as we know, this is the first report of organized gold nano-dot modified free-standing graphene as a SERS-active electrode.
 | ||
| Fig. 2 The AFM and TEM characterization of gold nano-dots obtained from AAO masks on different substrates. (a–c) Small gold nano-dots on silicon wafers, gold and graphene. The scale bar is 500 nm. (d) A 3D AFM image of the small gold nano-dots on graphene electrode. (e, f) The large and small gold nano-dots on free-standing graphene electrodes. The scale bar is 200 nm. | ||
In order to investigate the efficiency of the SERS substrate a standard analyte, Rhodamine 6G (R6G, 99% Sigma-Aldrich), was used to test the SERS-active substrate prior to the battery electrode testing. R6G ethanol solutions at different concentrations were prepared and then applied on to the SERS-active substrates. The results are shown in Fig. 3. In the initial test, the R6G solution with a concentration of 10−3 M was drop-cast on to the surface of the gold nano-dot modified silicon wafer. The Raman spectra of both the small gold nano-dot and large gold nano-dot modified silicon wafers show sharp peaks and identical patterns. In addition, the silicon wafer with the small gold nano-dots exhibits stronger peak intensities, indicating that the enhancement of the Raman signals of the small gold nano-dot modified substrate is greater than that of the large gold nano-dot modified substrate. Therefore, small gold nano-dots were selected to modify different substrates and study the SERS effect in the following work.
 | ||
| Fig. 3 Raman spectra of R6G on different SERS-active substrates. (a) Raman spectra of R6G (cast at a concentration of 10−3 M) on different sized gold nano-dot modified silicon wafer substrates. (b) Raman spectra of R6G (cast with different concentrations as noted) on a SERS-active (with small gold nano-dots) silicon wafer substrate. (c) Normalized Raman spectra of R6G (cast at a concentration of 10−6 M) on SERS-active (with small gold nano-dots) silicon wafer, gold and graphene substrates. (d) A series of Raman spectra collected along the dotted lines in (e) and (f). (e) Raman mapping of the peak intensity at a wavenumber of 1361 cm−1 on the SERS-active graphene substrate. (f) Raman mapping of the peak intensity at a wavenumber of 1650 cm−1 on the SERS-active graphene substrate. | ||
The SERS spectra with different concentrations of R6G are shown in Fig. 3b. When the R6G solution concentration decreases from 10−3 M to 10−6 M, the SERS substrate (small gold nano-dots on a silicon wafer) is still able to identify the analyte. To test the limit of the SERS substrate’s sensitivity, a monolayer of R6G molecules was prepared on the SERS substrate as follows:42–44 the SERS substrate was immersed in a 10−6 M R6G solution for two hours and then rinsed with ethanol. After this procedure, only a monolayer of R6G molecules was absorbed onto the surface.42–44 As shown in Fig. 3b (orange line), the Raman spectrum of the monolayer of R6G on the SERS-active electrode was obtained and found to match the standard pattern of R6G very well. In contrast, no Raman signal was observed for the bare silicon wafer, even when a high concentration (10−3 M) R6G solution was applied (Fig. 3b, black line).
Similarly, the SERS spectra of R6G on conductive substrates (small gold nano-dot modified gold and graphene) were studied. The results are shown in Fig. 3c. Compared with results from the SERS-active silicon wafer substrate, the SERS-active gold and graphene substrates also exhibited the capability to identify all R6G Raman peaks at a concentration of 10−6 M. The Raman spectra of unmodified graphene substrates are shown in Fig. S4, ESI.† The G-band (1590 cm−1)45 of graphene overlapped with part of the R6G spectrum. It is worth mentioning that the SERS-active substrate also enhanced the Raman signals from graphene.
One of the challenges for conventional SERS-active surfaces is that the active sites are distributed randomly on the SERS substrate. Previous work indicated that such a problem can be solved by applying regularly assembled nanoparticles on to the surface.34 In this work, the gold nano-dots were homogeneously deposited on the substrate. Hence, Raman mapping was carried out on the SERS substrate to confirm the uniform enhancement of Raman signals over the entire surface. In the Raman mapping experiment, the graphene substrate with small gold nano-dots was used and a R6G solution (10−6 M) was cast on to the substrate. An area of 20 × 20 μm was scanned using a Raman microscope. Fig. 3d shows the Raman spectra during the scan along the dashed lines indicated in Fig. 3e and f. Fig. 3e shows the contour information of the peak intensity at 1361 cm−1, where R6G has the strongest Raman signal. Fig. 3f shows the contour information of another representative R6G peak at 1650 cm−1. As shown in the mapping results, strong Raman signals are observed over the entire substrate. The uniform enhancement of the Raman signals is ideal for the study of intermediates/products on the SERS-active electrode.
SERS-active electrodes could be useful tools to analyze the products and intermediates formed on the battery electrode. As far as we knew, the only available reports of SERS-active electrodes for lithium–air batteries are based on roughened gold electrodes.18,26 In those reports, a pure gold cathode was studied. The information obtained from the roughened gold electrode was limited because the enhanced Raman signal was obtained only from the random “hot spots”. Thus in this work, SERS-active gold and graphene electrodes with Raman signal enhancement over the entire substrate are used in a lithium–air battery to study the materials formed during the discharge process.
The customized lithium–air battery in this research is illustrated in Fig. 4a. Although Swagelok-cells46 are typically used in the lab for lithium–air batteries, it was found that a three-electrode cell is easier to control since the working electrode needs to be examined under a Raman microscope frequently. In the three-electrode system, SERS-active electrodes (gold and graphene) were used as the working electrode. The electrolyte was 1 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) in dimethyl sulfoxide (DMSO). A picture of the device is shown in Fig. 4b. For comparison, a standard gold electrode was used as the working electrode to build a reference cell and this was measured as well.
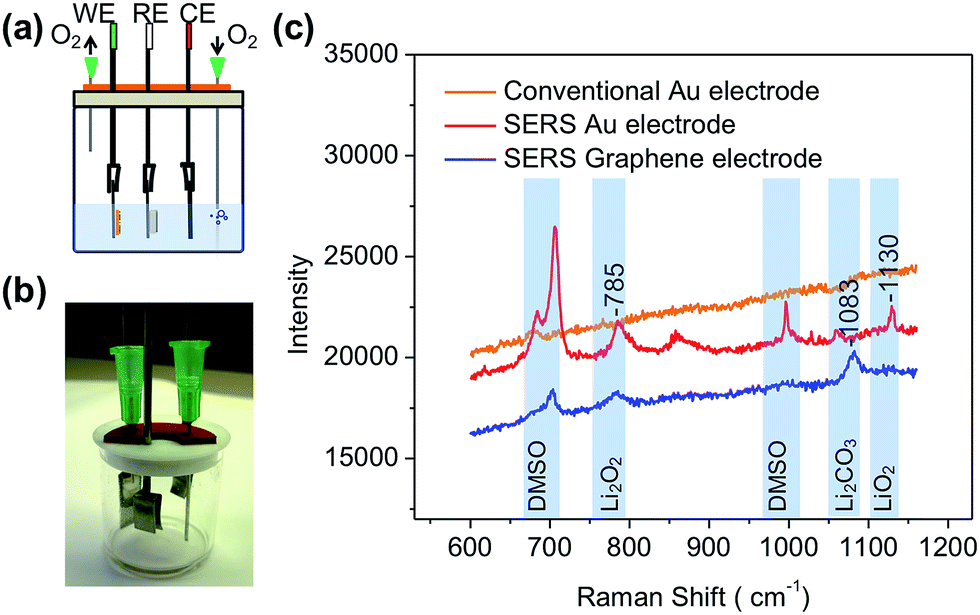 | ||
| Fig. 4 Fabrication and testing of the lithium–air battery with SERS cathodes. (a) Illustration of the battery device. SERS cathode is the working electrode (WE), Li foil and carbon cloth are used as the reference electrode (RE) and the counter electrode (CE), respectively. (b) A picture of the lithium–air battery device. (c) Raman spectra of different cathodes (a conventional gold electrode, the SERS-active gold electrode and the graphene electrode) after discharging. | ||
Cyclic voltammograms were recorded between 2.0 V and 3.8 V at a scan rate of 50 mV s−1 for the three-electrode cell. The results (ESI, Fig. S5†) show that the reduction peak is at around 2.4 V and the oxidation peak is at around 3.3 V, which is consistent with previous reports.47 A galvanostatic discharge experiment was conducted, and a discharge plateau at ∼2.7 V was observed at a discharge current density of 20 μA cm−2 (Fig. S6†). After discharge, the working electrode was transferred into a glovebox and washed with DMSO. The dried working electrode was sealed inside the glovebox for the Raman spectroscopy study. The Raman spectra of the discharged electrodes are shown in Fig. 4c. For the conventional gold electrode, the Raman spectrum shows no obvious signals because the amount of the discharging product is very limited on such a planar electrode. In contrast, sharp peaks are observed for the SERS-active gold electrode, including Li2O2 (785 cm−1) and LiO2 (1130 cm−1).26,48 There is one peak at 854 cm−1, which may be attributed to LiOH·H2O.49,50 When the SERS-active graphene electrode was used as a cathode, Li2O2 (785 cm−1) and Li2CO3 (1083 cm−1) were detected.26,48 These results provide important information for understanding the different reaction pathways of lithium–air batteries when different cathodes were used. Gold is a relatively stable cathode for lithium–air batteries.18 During the discharge process, the oxygen molecule will receive one electron to form superoxide anions (O2−) on the surface of the cathode. Then those superoxide anions react with Li+ to form LiO2. Recent research indicated that there are two possible reaction processes taking place after the LiO2 was formed on the electrode: (I) it can receive one more electron to form Li2O2 on the electrode surface;51 (II) with the existence of high donor number solvents such as DMSO, O2− and Li+ ion pairs can coordinate with solvent molecules and diffuse from the electrode surface to the electrolyte, where they follow a solution pathway (disproportionation) to form Li2O2.26 The detection of both LiO2 and Li2O2 on the SERS-active gold electrode suggests that the intermediate product (LiO2) can coexist with the final product (Li2O2) on the electrode after discharge.18,26 This result confirmed that gold is a stable electrode for both LiO2 and Li2O2. A small amount of LiOH·H2O was also detected on the SERS-active gold electrode. This can be explained as the result of the reaction between a proton and Li2O2/LiO2.14,52–57 The proton source can be residual water in the DMSO solvent. For the SERS-active graphene electrode, only Li2O2 and Li2CO3 are detected. These results indicated that intermediate lithium compounds (such as LiO2) may react with the carbon electrode to form Li2CO3 on the electrode surface.41,58 Although graphene itself is stable, the residual amorphous carbon or the terminal oxygen containing group may be reactive. The development of stable lithium–air batteries requires not only stable electrodes (or stabilized electrodes), but also a strategy to mitigate the side reactions associated with Li2O2 and LiO2.
Conclusions
In this work, a universal method to fabricate regularly patterned gold nano-dot modified SERS electrodes was developed. A free-standing AAO mask was used to generate well assembled nano-dots on silicon wafer, gold and graphene substrates. The substrates are SERS-active and able to detect a monolayer of R6G molecules. The conductive SERS-active substrates (gold and graphene) were used as cathodes to fabricate lithium–air batteries. The discharging products on the cathodes were investigated by SERS. The results indicated that the cathode reactions on these two electrodes are distinct from each other. The major products on the gold electrode are Li2O2, LiOH and LiO2. In contrast, the products on the graphene electrode are Li2O2 and Li2CO3. The SERS electrodes are so sensitive that even a small amount of product formed on the non-porous, planar electrode can be detected clearly. Such a property largely simplified the device fabrication for mechanistic studies. The SERS-active electrode in this work provided a uniform enhancement of the Raman spectra over the entire substrate, which is more suitable for detecting the intermediates and byproducts formed on the electrode. In addition, the SERS electrode preparation method in this work can be applied to any electrode for lithium–air batteries, rendering it a powerful tool in searching for novel electrode/catalysts for stable lithium–air batteries.Acknowledgements
The authors thank Dr B. Wang for help with SEM and TEM, and E. Laughlin for technical support. This work was supported by the National Science Foundation (NSF) through CBET 1505943, CBET 1336057, ECCS 1509754 and DMR 1554851, and the ACS Petroleum Research Fund (PRF# 54544-DNI 10). The authors are also grateful for financial support from the University of Akron.References
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- J. P. Zheng, R. Y. Liang, M. Hendrickson and E. J. Plichta, J. Electrochem. Soc., 2008, 155, A432–A437 CrossRef CAS.
- T. Zhang and H. Zhou, Nat. Commun., 2013, 4, 1817 CrossRef PubMed.
- R. Black, B. Adams and L. F. Nazar, Adv. Energy Mater., 2012, 2, 801–815 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Commun., 2012, 11, 19–29 CAS.
- R. Cao, J.-S. Lee, M. Liu and J. Cho, Adv. Energy Mater., 2012, 2, 816–829 CrossRef CAS.
- N.-S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y.-K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- J. Christensen, P. Albertus, R. S. Sanchez-Carrera, T. Lohmann, B. Kozinsky, R. Liedtke, J. Ahmed and A. Kojic, J. Electrochem. Soc., 2012, 159, R1–R30 CrossRef CAS.
- Y. Shao, F. Ding, J. Xiao, J. Zhang, W. Xu, S. Park, J.-G. Zhang, Y. Wang and J. Liu, Adv. Funct. Mater., 2013, 23, 987–1004 CrossRef CAS.
- S.-M. Xu, Q.-C. Zhu, J. Long, H.-H. Wang, X.-F. Xie, K.-X. Wang and J.-S. Chen, Adv. Funct. Mater., 2016, 26, 1365–1374 CrossRef CAS.
- Q.-C. Zhu, F.-H. Du, S.-M. Xu, Z.-K. Wang, K.-X. Wang and J.-S. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 3868–3873 CAS.
- Y. Qiao and S. Ye, J. Phys. Chem. C, 2016, 120, 8033–8047 CAS.
- W. Zhang, Y. Shen, D. Sun, Z. Huang, J. Zhou, H. Yan and Y. Huang, Nano Energy, 2016, 30, 43–51 CrossRef CAS.
- D. Sharon, M. Afri, M. Noked, A. Garsuch, A. A. Frimer and D. Aurbach, J. Phys. Chem. Lett., 2013, 4, 3115–3119 CrossRef CAS.
- B. D. McCloskey, A. Speidel, R. Scheffler, D. C. Miller, V. Viswanathan, J. S. Hummelshøj, J. K. Nørskov and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 997–1001 CrossRef CAS PubMed.
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS PubMed.
- B. D. McCloskey, D. S. Bethune, R. M. Shelby, G. Girishkumar and A. C. Luntz, J. Phys. Chem. Lett., 2011, 2, 1161–1166 CrossRef CAS PubMed.
- Z. Peng, S. A. Freunberger, L. J. Hardwick, Y. Chen, V. Giordani, F. Bardé, P. Novák, D. Graham, J.-M. Tarascon and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 6351–6355 CrossRef CAS PubMed.
- S. A. Freunberger, Y. Chen, N. E. Drewett, L. J. Hardwick, F. Barde and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 8609–8613 CrossRef CAS PubMed.
- A. Campion and P. Kambhampati, Chem. Soc. Rev., 1998, 27, 241–250 RSC.
- M. Moskovits, Rev. Mod. Phys., 1985, 57, 783–826 CrossRef CAS.
- A. Otto, I. Mrozek, H. Grabhorn and W. Akemann, J. Phys.: Condens. Matter, 1992, 4, 1143–1212 CrossRef CAS.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. R. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- F. S. Gittleson, W.-H. Ryu and A. D. Taylor, ACS Appl. Mater. Interfaces, 2014, 6, 19017–19025 CAS.
- Q. Yu and S. Ye, J. Phys. Chem. C, 2015, 119, 12236–12250 CAS.
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. A. Freunberger, P. C. Ashok, B. B. Praveen, K. Dholakia, J. M. Tarascon and P. G. Bruce, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS PubMed.
- Y. Chen, S. A. Freunberger, Z. Peng, O. Fontaine and P. G. Bruce, Nat. Chem., 2013, 5, 489–494 CrossRef CAS PubMed.
- C. Li, O. Fontaine, S. A. Freunberger, L. Johnson, S. Grugeon, S. Laruelle, P. G. Bruce and M. Armand, J. Phys. Chem. C, 2014, 118, 3393–3401 CAS.
- F. Tian, F. Bonnier, A. Casey, A. E. Shanahan and H. J. Byrne, Anal. Methods, 2014, 6, 9116–9123 RSC.
- W. Li, Y. Guo and P. Zhang, J. Phys. Chem. C, 2010, 114, 6413–6417 CAS.
- N. H. Kim and K. Kim, Chem. Phys. Lett., 2004, 393, 478–482 CrossRef CAS.
- F. Li, L. Zhang and R. M. Metzger, Chem. Mater., 1998, 10, 2470–2480 CrossRef CAS.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312–1314 CrossRef CAS PubMed.
- T. Kondo, H. Masuda and K. Nishio, J. Phys. Chem. C, 2013, 117, 2531–2534 CAS.
- X. Lang, T. Qiu, Y. Yin, F. Kong, L. Si, Q. Hao and P. K. Chu, Langmuir, 2012, 28, 8799–8803 CrossRef CAS PubMed.
- T. Qiu, W. Zhang and P. K. Chu, Phys. B, 2009, 404, 1523–1526 CrossRef CAS.
- M. Jung, S. K. Kim, S. Lee, J. H. Kim and D. H. Woo, J. Nanophotonics, 2013, 7, 073798 CrossRef.
- H. Masuda, H. Asoh, M. Watanabe, K. Nishio, M. Nakao and T. Tamamura, Adv. Mater., 2001, 13, 189–192 CrossRef CAS.
- M. Hideki and S. Masahiro, Jpn. J. Appl. Phys., Part 1, 1996, 35, L126 Search PubMed.
- M. Raoufi and H. Schönherr, Langmuir, 2014, 30, 1723–1728 CrossRef CAS PubMed.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, J. Am. Chem. Soc., 2013, 135, 494–500 CrossRef CAS PubMed.
- Y. Chen, Z. Li, Q. Xiang, Y. Wang, Z. Zhang and H. Duan, Nanotechnology, 2015, 26, 405301 CrossRef PubMed.
- G. Ritchie and E. Burstein, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 24, 4843–4846 CrossRef CAS.
- B. Kiraly, S. Yang and T. J. Huang, Nanotechnology, 2013, 24, 245704 CrossRef PubMed.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- Y.-M. Chen, A. Xie and Y. Zhu, ChemElectroChem, 2015, 2, 208–212 CrossRef CAS.
- F. Marchini, S. Herrera, W. Torres, A. Y. Tesio, F. J. Williams and E. J. Calvo, Langmuir, 2015, 31, 9236–9245 CrossRef CAS PubMed.
- S. Hy, F. Felix, J. Rick, W. N. Su and B. J. Hwang, J. Am. Chem. Soc., 2014, 136, 999–1007 CrossRef CAS PubMed.
- V. I. Tyutyunnik, J. Raman Spectrosc., 2000, 31, 559–563 CrossRef CAS.
- S. F. Parker, K. Refson, R. I. Bewley and G. Dent, J. Chem. Phys., 2011, 134, 084503 CrossRef PubMed.
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef CAS PubMed.
- T. Liu, M. Leskes, W. Yu, A. J. Moore, L. Zhou, P. M. Bayley, G. Kim and C. P. Grey, Science, 2015, 350, 530–533 CrossRef CAS PubMed.
- B. D. McCloskey, C. M. Burke, J. E. Nichols and S. E. Renfrew, Chem. Commun., 2015, 51, 12701–12715 RSC.
- D. G. Kwabi, T. P. Batcho, C. V. Amanchukwu, N. Ortiz-Vitoriano, P. Hammond, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. Lett., 2014, 5, 2850–2856 CrossRef CAS PubMed.
- B. D. McCloskey, A. Valery, A. C. Luntz, S. R. Gowda, G. M. Wallraff, J. M. Garcia, T. Mori and L. E. Krupp, J. Phys. Chem. Lett., 2013, 4, 2989–2993 CrossRef CAS PubMed.
- R. Younesi, P. Norby and T. Vegge, ECS Electrochem. Lett., 2014, 3, A15–A18 CrossRef CAS.
- N. Mozhzhukhina, L. P. Méndez De Leo and E. J. Calvo, J. Phys. Chem. C, 2013, 117, 18375–18380 CAS.
- D. M. Itkis, D. A. Semenenko, E. Y. Kataev, A. I. Belova, V. S. Neudachina, A. P. Sirotina, M. Hävecker, D. Teschner, A. Knop-Gericke, P. Dudin, A. Barinov, E. A. Goodilin, Y. Shao-Horn and L. V. Yashina, Nano Lett., 2013, 13, 4697–4701 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: SEM images of AAO templates and the as-prepared gold nano-dots. Height and morphology characterization of the gold nano-dots. Raman spectra of the graphene SERS electrode. Cyclic voltammograms and discharge profiles of a Li–O2 battery with the SERS electrode. See DOI: 10.1039/c6ra23331g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |