
The first computational study for the oxidative aromatization of pyrazolines and 1,4-dihydropyridines using 1,2,4-triazolinediones: an anomeric-based oxidation
Abstract
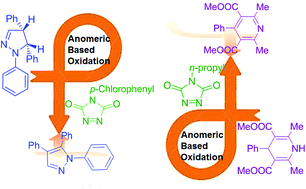
In this study, the oxidative aromatization process of 1,3,5-trisubstituted pyrazolines and 1,4 dihydropyridines, as pharmaceutically important structural motifs, using 1,2,4-triazolinediones as efficient oxidative agents were investigated. The achieved data from this computational study confirmed that the newly expanded term of “anomeric-based oxidation” for the aromatization of 1,3,5-trisubstituted pyrazolines and 1,4 dihydropyridine derivatives occurs by the common concerted oxidation via hydrogen abstraction–addition mechanism.
 Please wait while we load your content...
Please wait while we load your content...

