
Germanene: a new electronic gas sensing material
Abstract
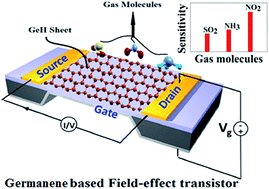
The structural stability and electronic properties of the adsorption characteristics of several toxic gas molecules (NH3, SO2 and NO2) on a hexagonal armchair of a germanene monolayer were investigated using density functional theory (DFT) based on an ab initio method. The sensitivity of the germanene monolayer has been investigated by considering the most stable adsorption configurations, adsorption energies, projected density of states and charge transfer between the monolayer and gas molecules. The adsorption energy of NO2 gas molecules on the germanene monolayer was the lowest energy (273.72 meV: B-type configuration) compared to all other possible configurations and also higher than that of NH3 and SO2. The charge transfer between the NO2 gas molecules and the germanene is the same order of magnitude, but larger than compared to that of NH3 and SO2 gas molecules. The higher charge transfer between the monolayer and gas molecules shows that this configuration can be utilized in germanene based field effect transistor (FET) sensors due to its greater stability and sensitivity.
 Please wait while we load your content...
Please wait while we load your content...

