
The design, synthesis and biological evaluation of pro-EGCG derivatives as novel anti-vitiligo agents†
Abstract
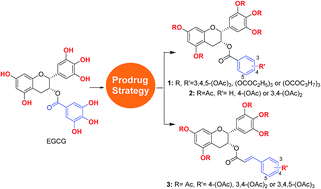
With the aim of overcoming the instability and poor membrane permeability of epigallocatechin-3-gallate (EGCG), a series of prodrugs of EGCG and its derivatives (pro-EGCGs) were designed and synthesized, and their protective effect on melanocytes against H2O2-induced cell damage was biologically evaluated. The enhanced potency of pro-EGCGs could be clearly observed, and the most potent compound, 3c, showed excellent protective effect of melanocytes against H2O2-induced cell death, and good effect on lactate dehydrogenase (LDH) release from melanocytes under oxidant stress. In addition, compounds 14b and 14c, which are the parent compounds of 3b and 3c, also showed good inhibitory activities against JAK1, JAK2 and JKA3, which are potential therapeutic targets for anti-vitiligo agents, demonstrating the promising perspective of 3b and 3c as both anti-oxidant and immuno-related agents for anti-vitiligo treatment.
 Please wait while we load your content...
Please wait while we load your content...

