
Screening PEGylated polyethylenimine derivatives for safe and efficient delivery of gene materials
Xuelan Tang†
a,
Ping Lu†b,
Mingfeng Qiu†a,
Jianjun Chenc,
Lin Mab,
Yanan Suna,
Feng Zhenga,
Enge Xua,
Jing Shengb and
Jing Su‡
*a
aSchool of Pharmacy, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: jingsu@sjtu.edu.cn; Fax: +86-21-34204052; Tel: +86-21-34204052
bShanghai Ninth People's Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200011, China
cDepartment of Pharmaceutical Sciences, College of Pharmacy, Chicago State University, Chicago, IL 60628, USA
First published on 2nd November 2016
Abstract
Gene-based therapy has broad and important clinical applications, and the non-viral vector delivery of exogenous nucleic acids is commonly used for gene therapy. Among the numerous gene delivery vectors, the cationic polymer vectors are considered to be the most promising materials for gene delivery and gene therapy. In this study, we chemically modified a novel cross-linked PEI derivative PEI–Et through PEGylation, as PEGylation can reduce the cytotoxicity of PEI–Et by shielding the excess positive surface charge of PEI–Et. To screen out an optimal molar ratio of PEI–Et to PEG for efficient delivery of gene materials, we first synthesized three kinds of PEG–Et cationic polymers with different molar ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1, 1:2) of PEI–Et to PEG, then we prepared and characterized the PEG–Et/DNA complexes and PEG–Et/siRNA complexes, and finally we tested the cytotoxicity and transfection efficiency of the PEG–Et/DNA complexes as well as the gene silencing efficiency of the PEG–Et/siRNA complexes. The results suggest that the gene vector PEG–Et 1:1 (with a 1:1 molar ratio of PEI–Et to PEG) was the best among the three types of PEG–Et (PEG–Et 1:1, PEG–Et 2:1, and PEG–Et 1:2) for condensing DNA and siRNA into nanoparticles, as it has significant lower cytotoxicity, higher gene transfection and siRNA silencing efficiency.
:1, 2:1, 1:2) of PEI–Et to PEG, then we prepared and characterized the PEG–Et/DNA complexes and PEG–Et/siRNA complexes, and finally we tested the cytotoxicity and transfection efficiency of the PEG–Et/DNA complexes as well as the gene silencing efficiency of the PEG–Et/siRNA complexes. The results suggest that the gene vector PEG–Et 1:1 (with a 1:1 molar ratio of PEI–Et to PEG) was the best among the three types of PEG–Et (PEG–Et 1:1, PEG–Et 2:1, and PEG–Et 1:2) for condensing DNA and siRNA into nanoparticles, as it has significant lower cytotoxicity, higher gene transfection and siRNA silencing efficiency.
1. Introduction
The exogenous nucleic acids such as DNA, mRNA, siRNA, miRNA and antisense oligonucleotides delivered by non-viral vectors are commonly used for gene-based therapy.1 Non-viral vector-based gene delivery systems have many advantages compared to viral vectors and some of them have entered clinical trials such as the new polymers and lipids.2–4 Nonetheless, the results from these clinical trials are not optimistic due to the presence of numerous barriers, for example, the delivery technology for gene-based therapy has become one of the most challenging hurdles to overcome, as the current non-viral vector-based gene delivery methods suffer from high cytotoxicity and low transfection efficiency. Therefore, there is a pressing need to develop a safe and efficient gene delivery system with minimum adverse effects. The cationic polymer based vectors as non-viral vectors have been developed as the most promising materials for gene delivery and gene-based therapy owing to several advantages, including high delivery efficiency, the ease of chemical modification, and the ability to deliver larger genetic payloads.5–7Polyethylenimine (PEI), especially PEI 25 kDa, has been successfully used as non-viral gene delivery carriers in both in vitro and in vivo studies with the characteristic known as the “proton sponge effect”.8 But PEI is non-degradable and is associated with cytotoxicity due to the excess positive surface charge. In order to reduce the cytotoxicity and improve the biodegradability, various PEIs have been modified into PEI polyplexes with biodegradable linkages, such as disulfide linkage,9 ester linkage,10,11 amide linkage,12 etc. These PEI-based polyplexes have a wide range of applications, for example, the polyplex ssPEI which was bound to SH–siRNA through a disulfide bridge has been used for efficient anti-cancer gene therapy.13 Another polyplex PEOz-PLA-g-PEI-SS also showed high efficacy by co-delivery of both DNA minicircles and doxorubicin.14 We previously designed and synthesized a degradable small-molecular-weight PEI derivative (PEI–Et), which was able to condense plasmid DNA into nanoparticles and exhibited significantly higher transfection efficiency compared to PEI 25 kDa.15 Nonetheless, the problem of cytotoxicity caused by the excess positive surface charge of PEI–Et has not been addressed. Therefore, after intravenous injection, the complexes may tend to form aggregates owing to charge interaction between the vectors and the serum components,16 which can cause toxicities.
Structural modifications can be done to reduce the toxicities caused by the positive surface charge of cationic polymers. One of the commonly used approaches is to modify the structure of the cationic polymer such as PEI by grafting a biocompatible polymer of neutral charge onto the PEI, as it can exert different effects on gene transfection17 according to the recent applications. PEG (polyethylene glycol) is a polymer with neutral charge and has been widely used for drug delivery purposes due to its high biocompatibility and benign toxicity profiles. The PEGylation to shield the excess positive surface charge of the PEI–Et complexes could help minimize the nonspecific interaction with serum components18 and prolong the systemic circulation time.19 By PEGylation of PEIs, various PEI-based polyplexes such as PEG–PCL–PEI,20 MoS2–PEI–PEG21 and PLLA–PEG–PLLA22 have been prepared for more efficient gene delivery.
In our research, we used PEGylation to make more accurate chemical design and to screen out an optimal molar ratio of PEI–Et to PEG in the PEGylated PEI–Et derivatives (PEG–Et) for efficient delivery of gene materials (Fig. 1A). We first precisely synthesized three kinds of PEG–Et cationic polymers with different molar ratio (1:1, 2:1, 1:2) of PEI–Et to PEG, and then the PEG–Et polymers were complexed with DNA or siRNA to generate the PEG–Et/DNA and PEG–Et/siRNA complexes which were characterized by agarose gel electrophoresis (AGE), particle size analysis, zeta potential, and transmission electron microscopy (TEM), and finally we tested the cytotoxicity and transfection efficiency of the PEG–Et/DNA complexes as well as the gene silencing efficiency of PEG–Et/siRNA complexes, to screen out the best molar ratio of PEI–Et to PEG in the PEGylated PEI–Et derivatives (PEG–Et) for condensing genes into nanoparticles.
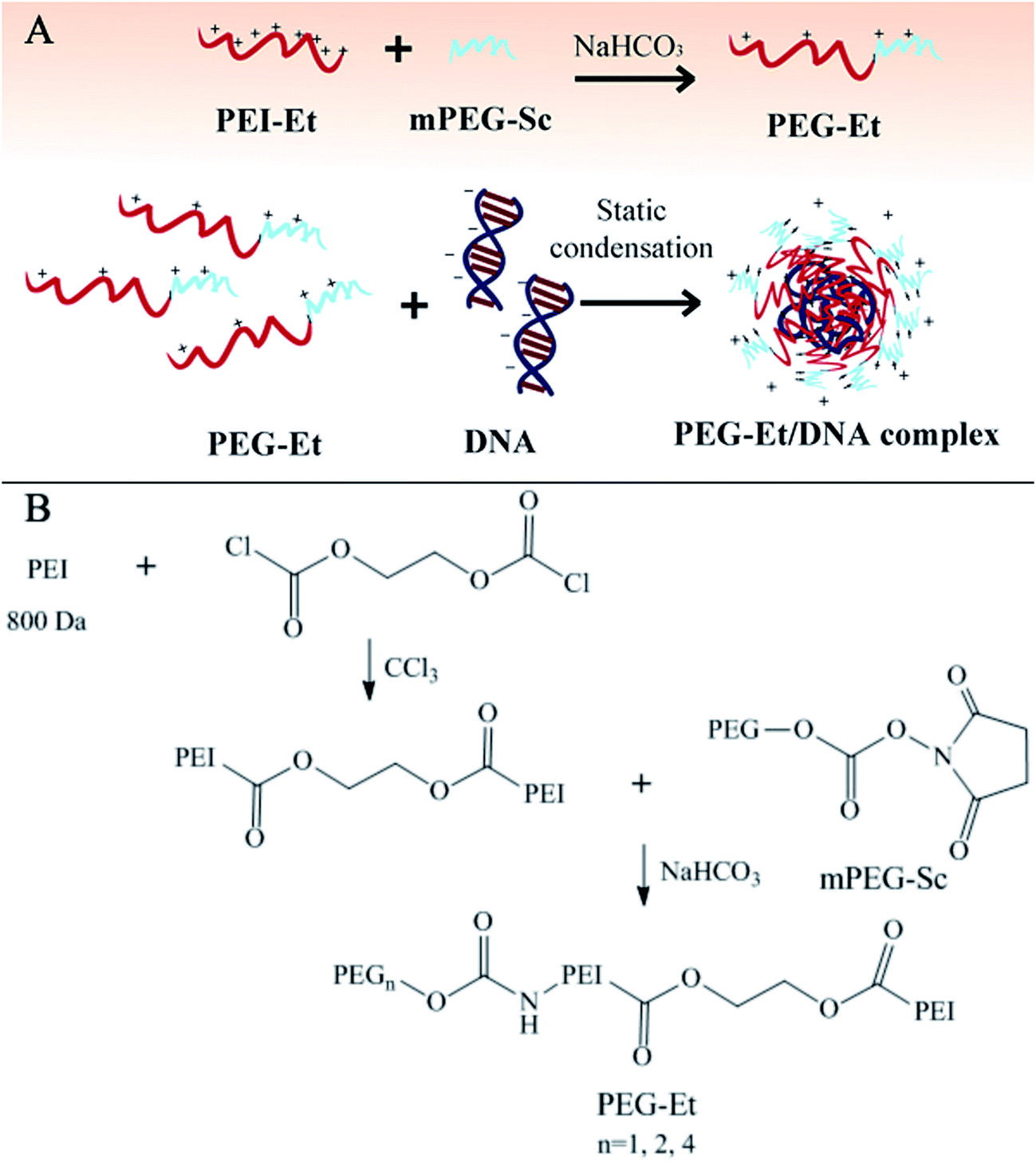 | ||
| Fig. 1 Preparation of PEG–Et/DNA complexes (A) and synthetic route of PEG–Et (B). | ||
2. Materials and methods
2.1. Materials
We derived branched PEI (25 kDa, 800 Da), ethylene bis(chloroformate), ethidium bromide (EB), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) from Sigma-Aldrich (St Louis, MO, USA), methoxy-poly(ethylene glycol)–succinimidyl carbonate (mPEG–Sc; Mw = 2000 Da) from Yare Biotech (Shanghai, China). We got MicroBCA protein assay kit from Pierce (Rockford, IL, USA), and obtained luciferase assay kit from Promega (Madison, WI). All other solvents and reagents were analytical grade. Dulbecco's Modified Eagle's Medium (DMEM), fetal bovine serum (FBS) and trypsin–EDTA were get from PAA (Cölbe, Germany). The siRNA used in the study were purchased from GenePharma (Shanghai, China).2.2. Cell culture
The cell lines CT-26 (colon adenocarcinoma cell), HeLa, MCF-7 (Michigan Cancer Foundation-7) and the cell lines SMMC7721 (human hepatic cancer cell) which could express luciferase protein steadily were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in DMEM medium and supplemented with 10% FBS, then maintained at 37 °C in a humidified incubator with 5% CO2.2.3. Synthesis of PEI–Et and PEG–Et
To synthesize PEI–Et, the reaction between PEI 800 Da and ethylene bis(chloroformate) was carried out according to the method that we have previously reported.15 Briefly, 0.04 mol of ethylene bis(chloroformate) solution (anhydrous chloroform) was slowly added to 0.06 mol of PEI 800 Da solution (anhydrous chloroform) with stirring in an ice bath and under nitrogen atmosphere for 24 hours. After removal of solvents, the sample was dissolved in water and dialyzed against distilled water for 2 days with a dialysis tube (MWCO: 3500 Da), the residue was subjected to lyophilization to give the polymer PEI–Et. The PEI–Et was then reserved at −20 °C for future use.The second step was the synthesis of PEG–Et 1:1 (Fig. 1B). Briefly, dissolving 0.04 mmol of PEI–Et in 0.1 M sodium bicarbonate, and then add 0.04 mmol of mPEG–Sc. Stirring the reaction mixture at room temperature for 4 h. Later dialyzing the raw product against distilled water with a dialysis tube (MWCO: 3500 Da) for 2 days, and final with lyophilization. PEG–Et 1:2 and PEG–Et 2:1 were also synthesized according to the same method with different molar ratio of PEI–Et to PEG (1:2, 2:1). The resulting polymers PEG–Et were then reserved at −20 °C for future use.
2.4. Characterization of PEG–Et
Proton nuclear magnetic resonance (1H-NMR) (Mercury Plus 400; Varian Inc., PaloAlto, CA) was used to estimate PEG–Et structure. 1H-NMR spectra of the polymers was recorded with a Varian Unity 300 MHz spectrometer (Varian Inc.), and D2O was used as a solvent. The molecular weight of PEG–Et was measured by GPC relative to PEG standards (Mw range Mp: 106, 430, 633, 1400, 4290, 7130, 12600, 20600 Da), with a Waters high-pressure liquid chromatography (HPLC) system (Milford, MA) that equipped with a gel permeation chromatography (GPC) and a refractive index detector. The mobile phase of HPLC was ultrapure water.
2.5. Preparation of PEG–Et/DNA complexes
PEG–Et/DNA complexes were freshly prepared before use. According to the different w/w ratio, PEG–Et and DNA were separately diluted in phosphate-buffered saline (PBS) to form the suitable concentration. Next, adding the polymer solutions to the DNA with gentle vortex to condense the complexes and then incubating for 30 minutes at room temperature prior to use.2.6. Gel retardation assay
Agarose gel electrophoresis (AGE) was used to evaluate the DNA condensation ability of the polymers. PEG–Et/DNA complexes were prepared at disparate w/w ratios (1, 3, 5, 10, 20 and 30). After incubation at room temperature for 30 minutes, the complexes were mixed with 2 μL of 5× gel-loading buffer and then were electrophoresed on 1% (w/v) agarose gels containing EB (0.5 μg mL−1) in 1× Tris-acetate (TAE) buffer at 110 V for 30 minutes. The locations of the DNA bands were visualized by using a UV illuminator (Tanon 2500, Shanghai).2.7. Particle size and zeta potential measurements
The particle size analyzer (Zetasizer Nano ZS90, Malvern, Britain) was used to measure the particle size and zeta potential of complexes. PEG–Et/DNA complexes were prepared at desired w/w ratios (1, 3, 5, 10, 20, 30 and 50). The measurement was carried out at 25 °C with the electric field strength of 7 V cm−1 and the scattering angle of 90 degree. Each sample was carried out in triplicate.2.8. Surface morphology
Transmission electron microscopy (TEM) was used to observe the morphology of complexes at w/w ratio 10. 20 μL of the complexes were placed on a copper grid (100 mesh), and then dried at room temperature. Then it was examined under 120 kV TEM (Tecnai G2 Spirit Biotwin, FEI, USA).2.9. Cytotoxicity assay
Cytotoxicity profiles of PEG–Et and PEG–Et/DNA complexes were measured by MTT assay in CT-26, HeLa and MCF-7 cell lines with PEI 25 kDa as a control. Cells were seeded in 96-well plates at an initial density of 5000 cells per well in 100 μL of DMEM containing 10% FBS. When the cells covered the plate, the media were replaced with polymers at diverse concentrations (1, 5, 10, 20, 50 and 100) or polymer/DNA complexes at different w/w ratios (3, 5, 10, 20, 30 and 50) which dissolved in fresh serum-free DMEM. After 4 hours' incubation, the media were removed, and 125 μL of stock solution of MTT (0.5 mg mL−1 in fresh serum-free DMEM) was added to each well. The cells were incubated for additional 6 hours, then, the media were replaced with dimethyl sulfoxide (DMSO) and 150 μL of DMSO was added to the well, the plates were gently shaken for 1 minute to ensure the complete dissolution of formazan. Finally, using an ELISA reader (SpectraMax M3, USA) to measure the absorbance at 570 nm (with 630 nm as a reference wavelength). The cell viability was represented as absorption percentage to that of the control test without polymers. Each sample was performed for six replicates.2.10. Gene transfection efficiency assay
PEG–Et-mediated gene transfection efficiency was evaluated in CT-26, HeLa and MCF-7 cell lines. Cells were seeded in 48-well plates at an initial density of 5 × 104 cells per well in 500 μL of DMEM containing 10% FBS. When the cells covered the plate, the cells were washed with PBS, and the media were exchanged for polymers at various concentrations or polymer/DNA complexes at various w/w ratios (3, 5, 10, 20, 30 and 50) which dissolved in fresh serum-free DMEM. Then it was incubated for another 4 hours. Cells transfected with PEI 25 kDa/DNA (optimal w/w) were used as positive control. Next, the media were replaced with 500 μL of fresh DMEM containing 10% FBS and incubated for 48 hours. Then the cells were added 75 μL of cell lysis buffer, and incubated for 30 minutes after washing with PBS. The luciferase assay was performed according to the instructions of manufacturer (Promega). The protein concentration of the cell lysates was determined with MicroBCA protein assay kit. The relative light units (RLU) of luciferase expression were determined along with a luminometer. The luciferase activity was expressed according to RLU mg−1 protein. Each sample was carried out in triplicate.2.11. Gene silencing efficiency assay
It is same as the method of gene transfection efficiency assay (2.10), the gene silencing was evaluated in SMMC7721 cell. Cells transfected with disordered siRNA were used as negative control, and cells transfected with PEI–Et/siRNA were used as positive control.2.12. Statistical analysis
Data were given as mean ± standard error (SE). All statistical analyses were performed with SPSS13.0 software. Student's t-test (two-tailed) was assigned to test the significance of the differences between two groups. The level of P < 0.05 was considered to represent significant difference and the level of P < 0.01 was considered to represent very significant difference.3. Results and discussion
3.1. Synthesis and characterization of polymers
PEG–Et was synthesized by the conjugation of mPEG to PEI–Et. The structure of PEG–Et was confirmed by 1H-NMR. The NMR spectrum of PEG–Et has the characteristic peaks for PEI–Et and mPEG (Fig. 2). The sharp “PEG peak” at 3.6 ppm (Fig. 2B–D) and the absence of this “PEG peak” in the NMR spectrum of PEI–Et (Fig. 2A) indicated that mPEG was chemically conjugated to PEI–Et successfully. A GPC instrument was used to detect the average molecular weight (Mw) of PEG–Et 1:1, PEG–Et 2:1, PEG–Et 1:2 as 4468, 5168, 3957 Da respectively (Table 1). The dispersity (D = Mw/Mn) was from 2.19 to 2.45 which inferred the branch structure of PEG–Ets. When the feed ratio of PEI–Et to PEG was 1:2, the lowest molecular weight PEG–Et 1:2 (Mw = 3957) was obtained which can be interpreted as too much PEG being linked to PEI–Et, because the feed ratio of PEI–Et to PEG was 1:2. The highest molecular weight PEG–Et 2:1 (Mw = 5168) was achieved by a 2:1 feed ratio of PEI–Et to PEG, which showed that the excessive amount of PEI–Et was linked to PEG. The medium molecular weight PEG–Et 1:1 (Mw = 4468) was acquired by a 1:1 feed ratio of PEI–Et to PEG. Collectively, these results confirmed that PEG–Et series were synthesized successfully.
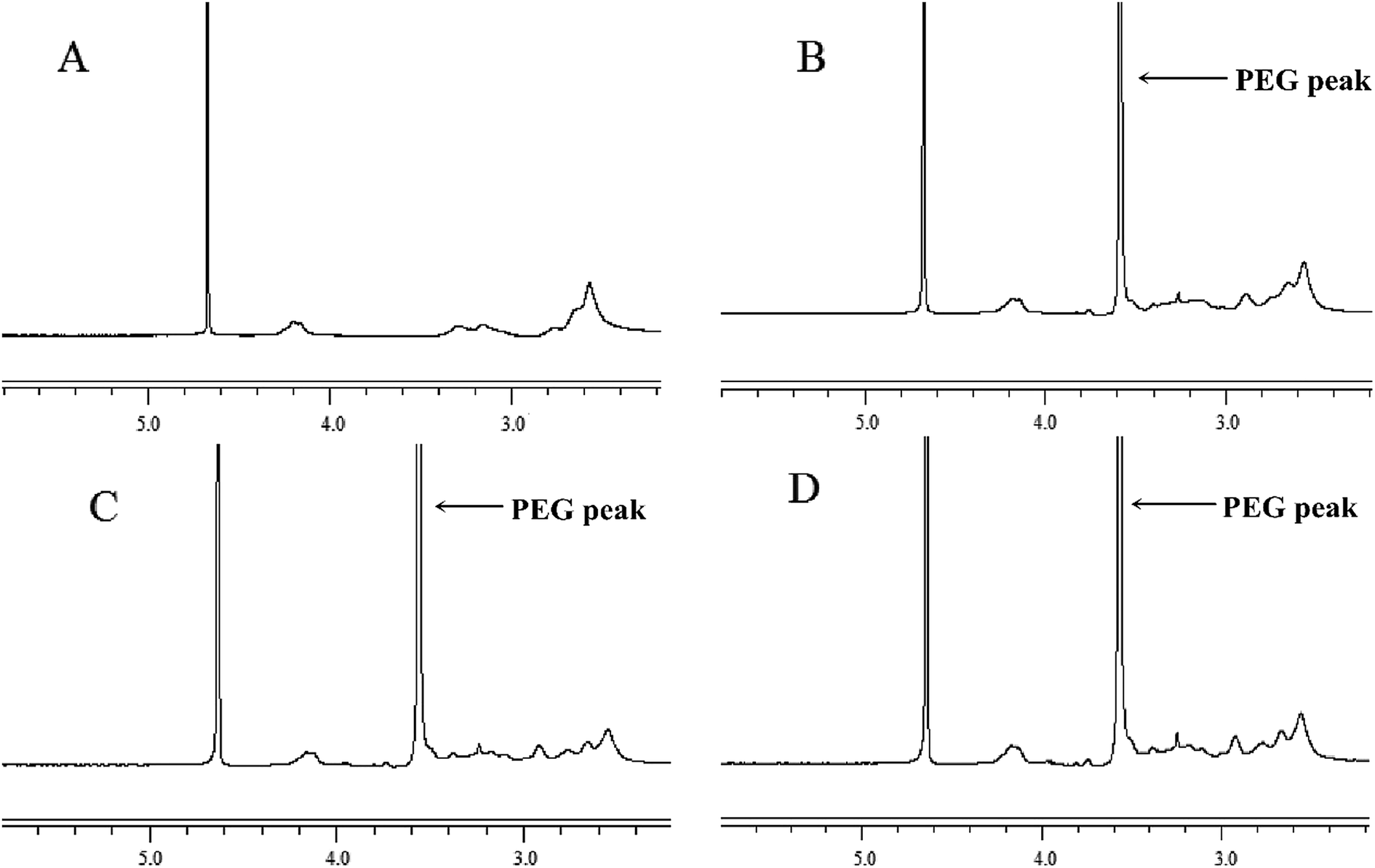 | ||
| Fig. 2 1H-NMR spectra of PEI–Et (A), PEG–Et 1:1 (B), PEG–Et 2:1 (C) and PEG–Et 1:2 (D). | ||
| Material | Mn (Da) | Mw (Da) | D |
|---|---|---|---|
| PEI–Et | 1281 | 2757 | 2.15 |
| PEG–Et 1:1 |
2043 | 4468 | 2.19 |
| PEG–Et 2:1 |
2343 | 5168 | 2.21 |
| PEG–Et 1:2 |
1612 | 3957 | 2.45 |
3.2. Characterization of PEG–Et/DNA complexes
:1 and PEG–Et 2:1 were successfully bound to DNA when the weight ratio (w/w ratio) was greater than 10, since the migration of DNA was retarded. PEG–Et 1:2 could not bind to DNA until the w/w ratio was greater than 20. We estimated that higher amount of PEG in PEG–Et 1:2 shielded more of the cationic surface charge of the polymers and lowered the DNA binding ability of PEG–Et 1:2, as the interaction of DNA with cationic polymers could protect the condensed DNA from enzymatic degradation, making it more effective for cellular transfection.24
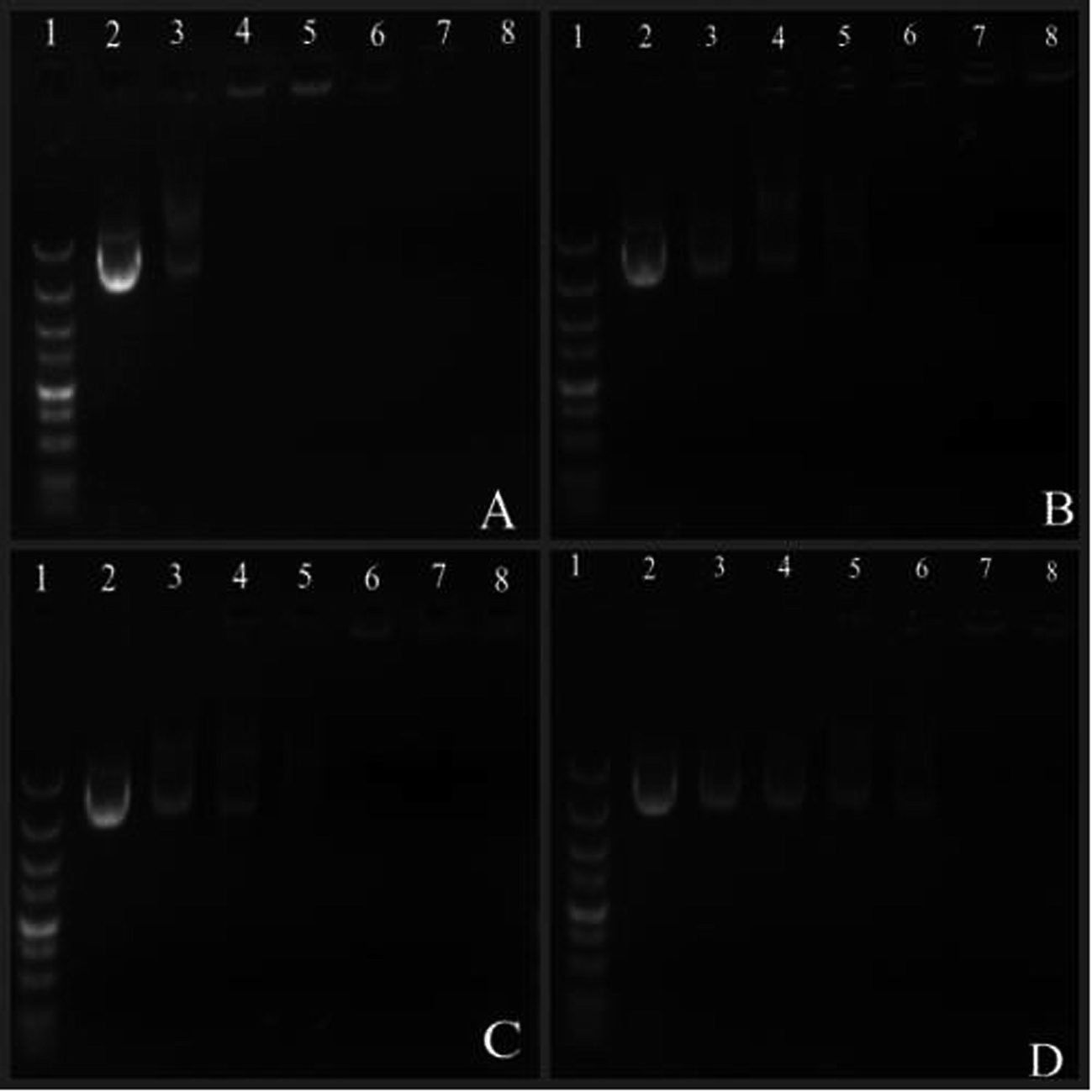 | ||
| Fig. 3 Agarose gel electrophoresis of polymer/DNA complexes at various w/w ratios. Notes: lane 1: marker, lane 2: naked DNA, lane 3–8: polymer/DNA complexes at w/w ratios of 1, 3, 5, 10, 20, 30 ((A) PEI–Et, (B) PEG–Et 1:1, (C) PEG–Et 2:1, (D) PEG–Et 1:2). | ||
To optimize the w/w ratio of PEG–Et to DNA, we monitored the size and zeta potential changes by varying the w/w ratios of PEG–Et to DNA. As the w/w ratios increase, the particle sizes of nanoparticles became stabilized (Fig. 4). When the w/w ratio was greater than 10, the particle size of PEG–Et 1:1/DNA was 120–140 nm, that of PEG–Et 2:1/DNA was 110–120 nm, and that of PEG–Et 1:2/DNA was bigger than 130 nm. In contrast to unPEGylated PEI–Et, the zeta potential of PEG–Et was relatively low due to the presence of the PEG group. The particle size of nanoparticles was found to be dependent on PEI concentration, and the concentration of PEI in nanoparticles also influenced the surface charge of nanoparticles.26 As shown in Fig. 4, when the weight ratio was greater than 10, the surface charge of PEG–Et 1:1/DNA was 3–6 mV, that of PEG–Et 2:1/DNA was 5–9 mV, and that of PEG–Et 1:2/DNA was lower than 1 mV. The zeta potential of PEI–Et was 9–14 mV. It is obvious that the surface charge of the complexes decreases with the extent of PEGylation. It is known that the high positive surface charge on the complexes could cause cationic cytotoxicity. Furthermore, under in vivo conditions, higher cationic charge could enhance nonspecific binding of the complexes to cellular components in the blood, erythrocytes, and/or endothelium cells in vessel walls.27 The size of the nanoparticle is a crucial factor in determining the rate of cellular uptake of nucleic acid polyplexes.28 Small size and weak positive charge are preferable for optimal cellular uptake of nanoparticles. For efficient gene delivery, a balance between the particle size and zeta potential should be considered. Though zeta potential of PEG–Et 1:2/DNA is low, which may decrease cytotoxicity, the particle size of PEG–Et 1:2/DNA is bigger than 200 nm when the w/w was 20, which is not suitable for cellular uptake. For PEG–Et 2:1/DNA polyplexes, the size of the nanoparticles is favorable (110–120 nm). However, the zeta potential (5–9 mV) may be high compare with that (3–6 mV) of PEG–Et 1:1/DNA polyplexes and should be addressed for delivery priority. We noticed the particle size of PEG–Et 1:1/DNA was a little higher (120–140 nm), but the zeta potential was quite smaller (3–6 mV) compared with PEI–Et/DNA. Therefore, we concluded that PEG–Et 1:1 is a promising delivery material based on its lower cytotoxicity, higher gene transfection efficiency and siRNA silencing efficiency in comparison to other polymers (PEI–Et, PEG–Et 1:2 and PEG–Et 2:1).
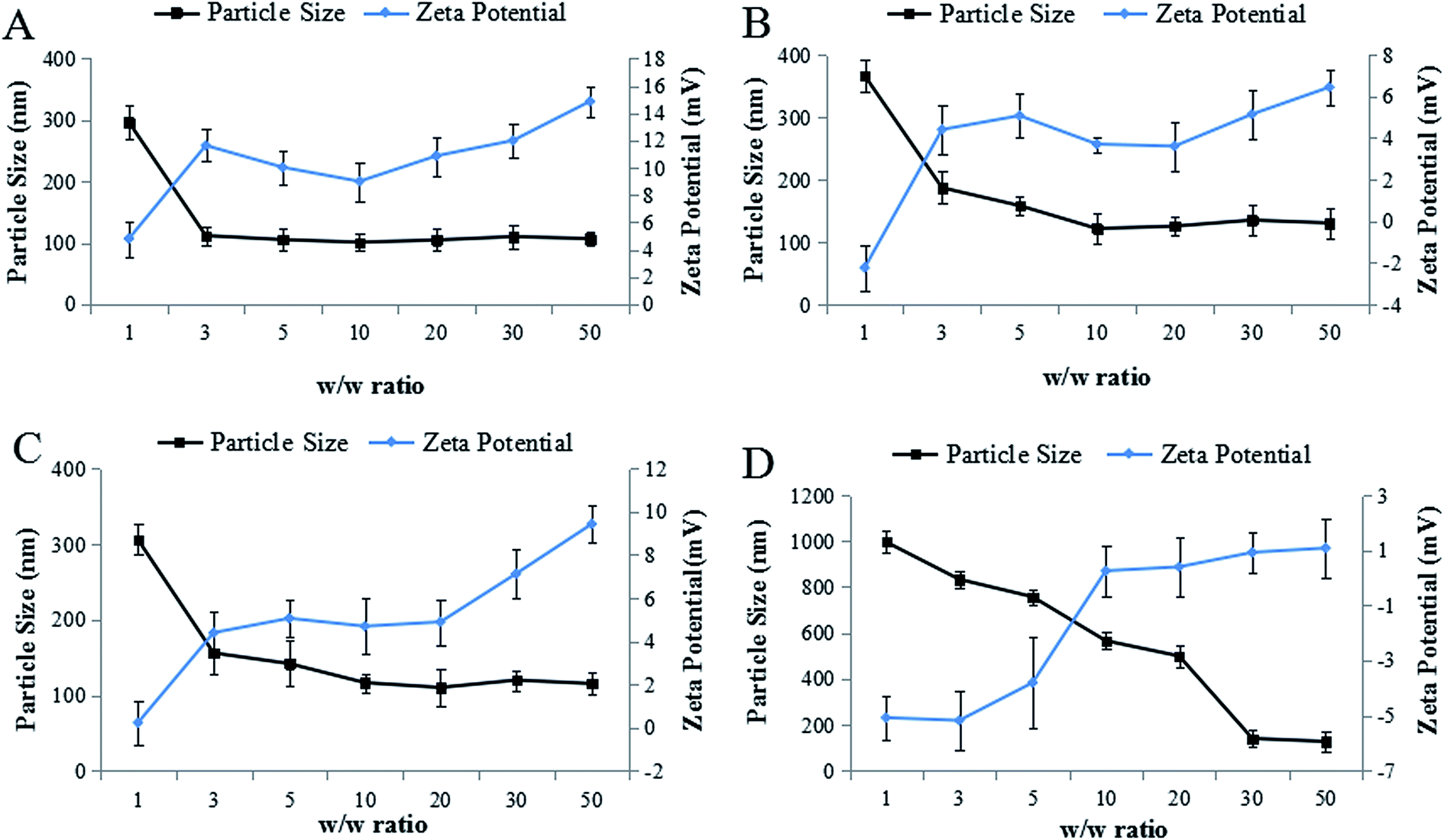 | ||
| Fig. 4 Particle size and zeta potential of polymer/DNA complexes at various w/w ratios (n = 3, (A) PEI–Et, (B) PEG–Et 1:1, (C) PEG–Et 2:1, (D) PEG–Et 1:2). | ||
:1. Fig. 5 showed the representative surface morphologies of PEG–Et/DNA complexes under TEM. The results indicated that PEG–Et 1:1/DNA, PEG–Et 2:1/DNA, PEI–Et/DNA were spherically shaped with a diameter of approximately 100 nm, which was in agreement with the particle size analysis using the Zeta-Plus instrument. However, the diameter of PEG–Et 1:2/DNA was over 500 nm, which was consistent with previous molecule weight analysis, gel retardation assay and DLS particle size measurement.
 | ||
| Fig. 5 Representative transmission electron microscopy image of polymer/DNA complexes at a w/w ratio of 20 ((A) PEI–Et, (B) PEG–Et 1:1, (C) PEG–Et 2:1, (D) PEG–Et 1:2). | ||
3.3. Cytotoxicity assay
Cytotoxicity is a primary obstacle for clinic applications of polycationic gene carriers. Low cytotoxicity is a prerequisite for the delivery of DNA materials through vectors. There are at least two types of cytotoxicity that mediated by PEG–Et: one direct toxicity associated with free PEG–Et and the other delayed toxicity associated with PEG–Et/DNA complexes.29 In this study, we measured the cytotoxicity of free polymers and polymer/DNA complexes. Free polymers were used to simulate a worst case scenario and to obtain results with larger sensitivity, since cytotoxicity was reduced when polymer/DNA complexes were formed.30 In vitro cytotoxicity was analyzed by MTT assay in CT-26, HeLa, and MCF-7 cell lines.Fig. 6 showed that the cytotoxicity of free polymers (Fig. 6A–C) and polymer/DNA complexes (Fig. 6D–F) increased with the increase of the polymer concentrations and w/w ratios in the three cell lines (HeLa, MCF-7 and CT-26 cells). As indicated in Fig. 6A–C, when the polymer concentration was 1 μg mL−1, all polymers (PEG–Ets, PEI–Et and PEI 25 kDa) showed almost 100% cell viability. As the polymer concentration was increased (5 μg mL−1 and 10 μg mL−1), cell viability of PEI 25 kDa slightly decreased, while PEG–Ets and PEI–Et still produced no cytotoxicity. When the polymer concentration was further increased to 50 μg mL−1, cell viability of PEI 25 kDa and PEI–Et dramatically decreased to 40.0% ± 1.7%, 27.9% ± 1.7%, 42.9% ± 2.0% and 65.4% ± 2.1%, 61.4% ± 1.8%, 70.5% ± 1.1%, but cell viability of PEG–Et 1:1 and PEG–Et 1:2 decreased very slightly to 90.2% ± 1.2%, 89.3% ± 1.5%, 92.7% ± 1.5% and 93.1% ± 1.7%, 91.5% ± 2.1%, 93.6% ± 1.9% in the three cell lines (HeLa, MCF-7 and CT-26 cells, respectively). Even when the polymer concentration was increased to 100 μg mL−1, the cell viabilities of PEG–Et 1:1 and PEI–Et 1:2 still exceed 60%, which is significantly higher than that of PEI–Et. In summary, for the cell viabilities of free polymers, PEG–Et 1:1 and PEG–Et 1:2 exhibited much lower cytotoxicity than that of PEI 25 kDa, PEI–Et, and PEG–Et 2:1 (the difference in cell viability is statistically significant, P < 0.01). We also tested the cell viability of polymer/DNA complexes with different w/w ratios in the three cell lines (HeLa, MCF-7 and CT-26 cells). As shown in Fig. 6D–F, when the w/w ratio was 3, all the polymer/DNA complexes showed 100% cell viability and exhibited no cytotoxicity. When the w/w ratio was 5, cell viability of PEI 25 kDa/DNA slightly decreased to 85.7% ± 2.0%, 88.6% ± 1.5%, 91.4% ± 2.0% (HeLa, MCF-7 and CT-26 respectively), our polymer (PEG–Ets or PEI–Et)/DNA complexes showed almost 100% cell viability. When we raised w/w ratio from 10 to 30, the cell viability of PEI 25 kDa/DNA drastically fell to 29.2% ± 1.2%, 41.6% ± 1.2%, 42.4% ± 1.6% (HeLa, MCF-7 and CT-26 respectively). However, PEG–Et 1:1 or PEG–Et 1:2/DNA complexes displayed over 80% cell viability which is significantly higher than that of PEI–Et/DNA or PEG–Et 2:1. When the w/w ratio was further increased to 50, the cell viability of PEG–Et 1:1 or PEG–Et 1:2/DNA complexes was over 70%. In conclusion, for the cell viabilities of polymer/DNA complexes, PEG–Et 1:1 and PEG–Et 1:2/DNA complexes displayed remarkably lower cytotoxicity than PEI–Et and PEG–Et 2:1/DNA complexes among the three cell lines.
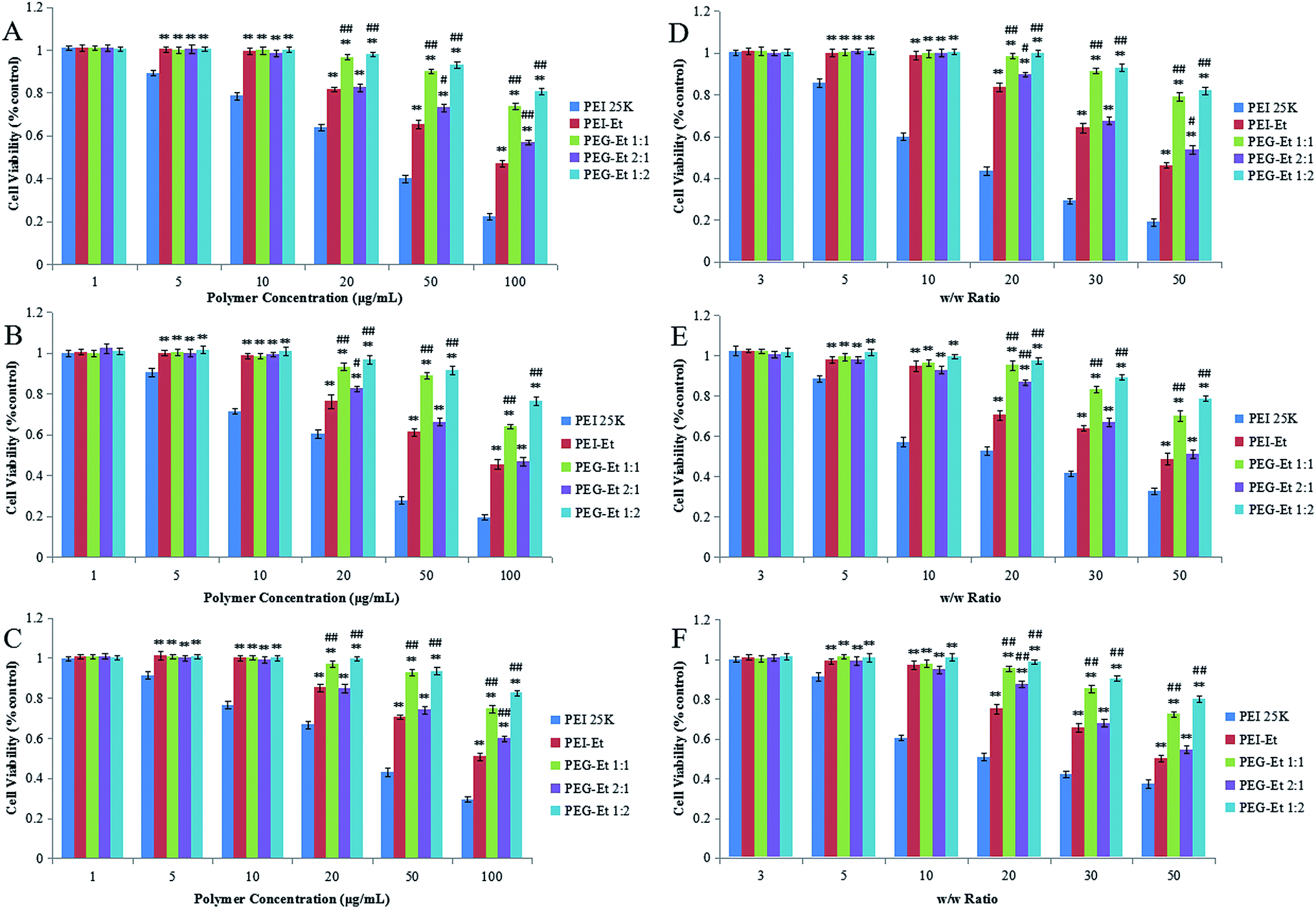 | ||
| Fig. 6 Cytotoxicity of the polymers at various concentrations and cytotoxicity of the polymer/DNA complexes at various w/w ratios in HeLa (A, D), MCF-7 (B, E) and CT-26 (C, F) cell lines (n = 6, ##P < 0.01, #P < 0.05 vs. PEI–Et; **P < 0.01 vs. PEI 25 kDa). | ||
Due to strong electrostatic interactions, the cytotoxicity of cationic polymers may be caused by the polymer aggregation on the cell surface with the plasma membrane, which could damage the cell membrane function.31 The PEG–Ets, especially PEG–Et 1:1 and PEG–Et 1:2, showed significantly lower cytotoxicity compared with PEI–Et. The higher cell viability of PEG–Et 1:1 and PEG–Et 1:2 was attributed to the higher constituents of PEG moieties which resulted in a weaker positive charge compared with that of PEG–Et 2:1, which was consistent with previous zeta potential analysis. We confirmed that after chemical modification of PEI–Et through PEGylation, PEG–Et 1:1 was selected to be the best system for further gene delivery study.
3.4. Gene transfection efficiency assay and gene silencing assay
Luciferase activity assays were performed in order to study the gene transfection efficiency of PEG–Et in vitro. PEI–Et and PEI 25 kDa at the optimal w/w ratio were used as positive control. Fig. 7 showed that the gene transfection efficiency of polymers increased with the increasing w/w ratios at a w/w ratio of <20, and then decreased at higher w/w ratios. Moreover, the transfection efficiency of PEG–Et 1:1 was the highest at a w/w ratio 20. As shown in Fig. 7D–F, when the w/w ratios were 10, 20 (HeLa, MCF-7 and CT-26 cells), 30 (MCF-7 cells), the transfection efficiency of PEG–Et 1:1 was higher than that of PEI 25 kDa, and the result was statistically significant with the level of P < 0.01. When the w/w ratios were 10, 20, the transfection efficiency of PEG–Et 1:1 was higher than that of PEI–Et, and the difference is significant with the level of P < 0.01 (MCF-7 and CT-26 cells) and P < 0.05 (HeLa cells). When the w/w ratio was 20, the transfection efficiency of PEG–Et 1:1 was 1.745 × 108 RLU mg−1, 2.695 × 108 RLU mg−1, 3.445 × 108 RLU mg−1 (HeLa, MCF-7 and CT-26 cells, respectively), and that of PEI–Et was 8.753 × 107 RLU mg−1, 9.345 × 107 RLU mg−1, 1.345 × 108 RLU mg−1. Therefore, the transfection efficiency of PEG–Et 1:1 was 1.99, 2.88, and 2.56 times higher than that of PEI–Et in HeLa, MCF-7, and CT-26 cells respectively. It was apparent that PEG–Et 1:1 was a better carrier than other polymers.
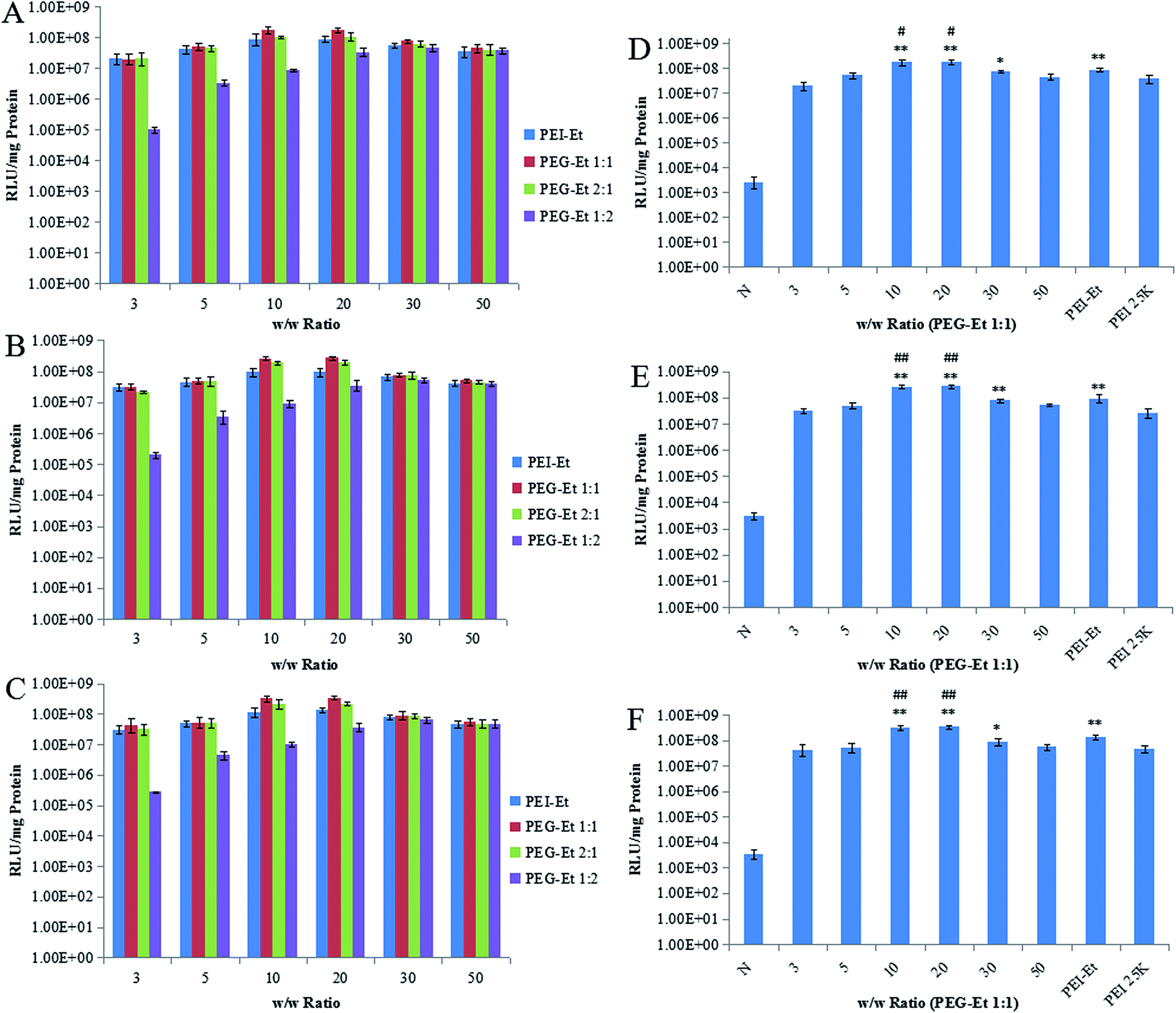 | ||
| Fig. 7 Transfection efficiency of polymer/PGL3DNA complexes at various w/w ratios in HeLa (A, D), MCF-7 (B, E) and CT-26 (C, F) cell lines in serum-free medium (n = 3, ##P < 0.01, #P < 0.05 vs. PEI–Et; **P < 0.01, *P < 0.05 vs. PEI 25 kDa, note: N: naked DNA). | ||
With the recent development of nanoparticles and conjugate formulations, RNAi-based therapies have gradually shown promising results.1 And RNA-induced silencing complex (RISC) of PEG–Et/siRNA complexes was also studied by luciferase activity assays. PEI–Et was used as a positive control. Fig. 8 showed that the gene silencing efficiency of PEG–Et increased at a w/w ratio of <20, and then decreased at higher w/w ratios. When the w/w ratios were 10, 20 and 30, the gene silencing of PEG–Et 1:1 was higher than that of PEI–Et (P < 0.01). The highest gene silencing of PEG–Et 1:1 was 50.1% ± 1.2% when the w/w ratio was 20. The gene silencing efficiency of the optimal weight ratio of PEI–Et was 42.5% ± 0.9%. Based on these results, it was apparent that PEG–Et 1:1 was a better carrier in comparison with PEI–Et.
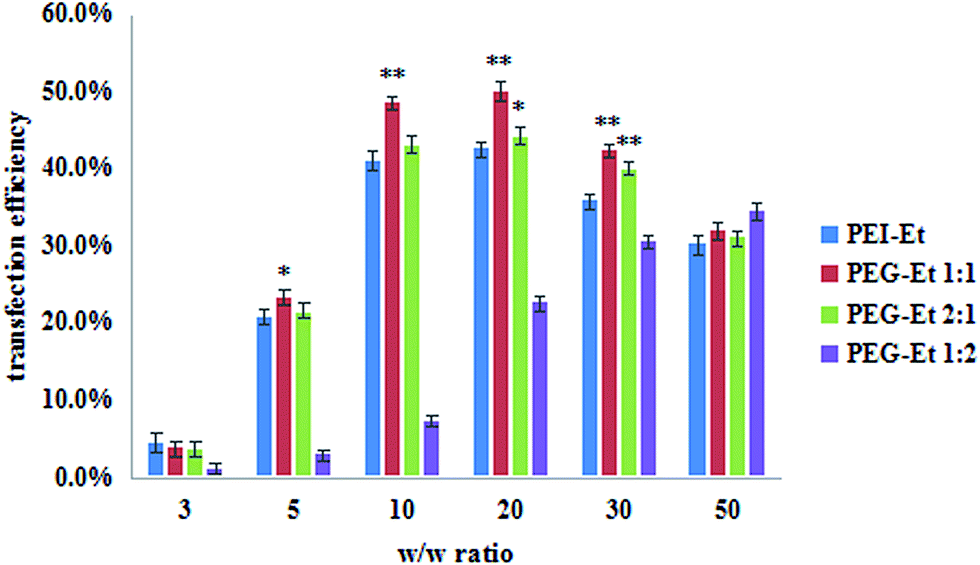 | ||
| Fig. 8 Gene silencing efficiency of PEG–Et/siRNA complexes at various w/w ratios in SMMC7721 cell lines in comparison with that of PEI–Et (n = 3, PEI–Et; **P < 0.01, *P < 0.05 vs. PEI–Et). | ||
These phenomena could be explained by the fact that a low w/w ratio would yield physically unstable complexes and poor transfection, whereas a high w/w ratio resulted in poor gene silencing due to their high stability, thus the gene materials could not be released from the complexes.32 Many reports suggested that the higher gene silencing efficiency of polyplexes might be attributed to the higher buffering capacity of these complexes and rapid unpackaging of nucleic acids from polymers when they are transfected into the cells.33–35 And PEGylation could shield excess positive surface charge and improve the survival rate of cells.36,37 Therefore, PEG–Et 1:1 had higher transfection efficiency and silencing efficiency.
4. Conclusion
In this paper, PEGylation of cationic polymers (PEI–Et) was explored to reduce the cytotoxicity of the polymers by shielding the excess positive surface charge of PEI–Et. Three types of PEGylated PEI–Ets were synthesized and characterized, namely, PEG–Et 1:1, PEG–Et 2:1, and PEG–Et 1:2. The PEG–Ets were subsequently complexed with DNA or siRNA to produce their PEG–Et/DNA or PEG–Et/siRNA derivatives. The particle size, zeta potential, and transmission electron microscopy of PEG–Et/DNA complexes were measured. The results indicated that the nanoparticles were stable, spheriform and about 100 nm in size with a surface charge of approximately 6 mV. Then a series of biological experiments were performed to investigate the cytotoxicity and gene transfection efficiency of the PEG–Et/DNA complexes at different w/w ratios, and the gene silencing efficiency of the PEG–Et/siRNA complexes. PEG–Et 1:1 was found to be the best among the three types of PEG–Ets (PEG–Et 1:1, PEG–Et 1:2, and PEG–Et 2:1) for efficient delivery of gene materials, as demonstrated by its higher gene transfection efficiency and gene silencing efficiency when compared to the other two types of PEG–Et/DNA complexes (PEG–Et 1:2 and PEG–Et 2:1) and PEI–Et. In addition, PEG–Et 1:1 also exhibited significantly lower cytotoxicity in vitro than PEI–Et and PEG–Et 2:1 due to the shielding of the positive surface charges of the polymers. Taken together, these findings suggested that PEG–Et 1:1 has the greatest gene transfection/silencing efficiency and minimum cytotoxicity among the three polymers. In summary, our research revealed that precisely chemical synthesis of PEGylated PEI derivatives (PEG–Ets) could provide the rationale of screening cationic degradable polymers for efficient delivery of gene materials. For our further study, we intend to investigate the effects of PEG–Et on gene expression in vivo, and the targeted delivery of gene materials by PEG–Et with further structural modifications.
Live subject statement
We declare that the live subjects (PEG–Et/DNA) in the manuscript were synthesized by ourselves, and the cell lines CT-26 (colon adenocarcinoma cell), HeLa, MCF-7 (Michigan Cancer Foundation-7) and the cell lines SMMC7721 (human hepatic cancer cell) were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All of our experiments were carried out at the School of Pharmacy, Shanghai Jiao Tong University.Acknowledgements
This work was financially supported by the Morning Star Plan (B) of SJTU (AF1700016), the National Natural Science Foundation of China (81977757, 81300092) and Shanghai Science and Technology Commission Funding (13401900801, 15401901700).References
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- M. E. Davis, Mol. Pharm., 2009, 6, 659–668 CrossRef CAS PubMed.
- K. T. Love, K. P. Mahon, C. G. Levins, K. A. Whitehead, W. Querbes, J. R. Dorkin, J. Qin, W. Cantley, L. L. Qin, T. Racie, M. Frank-Kamenetsky, K. N. Yip, R. Alvarez, D. W. Sah, A. de Fougerolles, K. Fitzgerald, V. Koteliansky, A. Akinc, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1864–1869 CrossRef CAS PubMed.
- S. C. Semple, A. Akinc, J. Chen, A. P. Sandhu, B. L. Mui, C. K. Cho, D. W. Sah, D. Stebbing, E. J. Crosley, E. Yaworski, I. M. Hafez, J. R. Dorkin, J. Qin, K. Lam, K. G. Rajeev, K. F. Wong, L. B. Jeffs, L. Nechev, M. L. Eisenhardt, M. Jayaraman, M. Kazem, M. A. Maier, M. Srinivasulu, M. J. Weinstein, Q. Chen, R. Alvarez, S. A. Barros, S. De, S. K. Klimuk, T. Borland, V. Kosovrasti, W. L. Cantley, Y. K. Tam, M. Manoharan, M. A. Ciufolini, M. A. Tracy, A. de Fougerolles, I. MacLachlan, P. R. Cullis, T. D. Madden and M. J. Hope, Nat. Biotechnol., 2010, 28, 172–176 CrossRef CAS PubMed.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS PubMed.
- H. Yu and E. Wagner, Curr. Opin. Mol. Ther., 2009, 11, 165–178 CAS.
- L. Jin, X. Zeng, M. Liu, Y. Deng and N. He, Theranostics, 2014, 4, 240–255 CrossRef PubMed.
- D. Sutton, S. Kim, X. Shuai, K. Leskov, J. T. Marques, B. R. Williams, D. A. Boothman and J. Gao, Int. J. Nanomed., 2006, 1, 155–162 CrossRef CAS PubMed.
- S. Taranejoo, J. Liu, P. Verma and K. Hourigan, J. Appl. Polym. Sci., 2015 DOI:10.1002/app.4209610.1002/app.42096.
- Q. F. Guo, T. T. Liu, X. Yan, X. H. Wang, S. Shi, F. Luo and Z. Y. Qian, Int. J. Nanomed., 2011, 6, 1641–1649 CAS.
- M. Thomas, Q. Ge, J. J. Lu, J. Z. Chen and A. M. Klibanov, Pharm. Res., 2005, 22, 373–380 CrossRef CAS.
- D. W. Dong, S. W. Tong and X. R. Qi, J. Biomed. Mater. Res., Part A, 2013, 101, 1336–1344 CrossRef PubMed.
- M. Muthiah, H. L. Che, S. Kalash, J. Jo, S. Y. Choi, W. J. Kim, C. S. Cho, J. Y. Lee and I. K. Park, Colloids Surf., B, 2015, 126, 322–327 CrossRef CAS PubMed.
- V. M. Gaspar, P. Baril, E. C. Costa, D. de Melo-Diogo, F. Foucher, J. A. Queiroz, F. Sousa, C. Pichon and I. J. Correia, J. Controlled Release, 2015, 213, 175–191 CrossRef CAS PubMed.
- Y. Q. Wang, J. Su, F. Wu, P. Lu, L. F. Yuan, W. E. Yuan, J. Sheng and T. Jin, Int. J. Nanomed., 2012, 7, 693–704 CAS.
- W. C. Tseng, L. Y. Su and T. Y. Fang, J. Biomed. Mater. Res., Part B, 2013, 101, 375–386 CrossRef PubMed.
- A. Malek, F. Czubayk and A. Aigner, J. Drug Targeting, 2008, 16, 124–139 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharm., 2008, 5, 505–515 CrossRef CAS PubMed.
- F. M. Veronese and G. Pasut, Drug Discovery, 2005, 10, 1451–1458 CrossRef CAS.
- T. Endres, M. Zheng, A. Kilic, A. Turowska, M. Beck-Broichsitter, H. Renz, O. M. Merkel and T. Kissel, Mol. Pharm., 2014, 11, 1273–1281 CrossRef CAS PubMed.
- J. Kim, H. Kim and W. J. Kim, Small, 2015, 1–9, DOI:10.1002/smll.201501655.
- D. G. Abebe, R. Kandil, T. Kraus, M. Elsayed, O. M. Merkel and T. Fujiwara, Macromol. Biosci., 2015, 15, 698–711 CrossRef CAS PubMed.
- R. Jiang, X. Lu, M. Yang, W. Deng, Q. Fan and W. Huang, Biomacromolecules, 2013, 14, 3643–3652 CrossRef CAS PubMed.
- S. C. Park, J. P. Nam, Y. M. Kim, J. H. Kim, J. W. Nah and M. K. Jang, Int. J. Nanomed., 2013, 8, 3663–3677 Search PubMed.
- W. Fan, X. Wu, B. Ding, J. Gao, Z. Cai, W. Zhang, D. Yin, X. Wang, Q. Zhu, J. Liu, X. Ding and S. Gao, Int. J. Nanomed., 2012, 7, 1127–1138 CAS.
- N. Kim, D. Jiang, A. M. Jacobi, K. A. Lennox, S. D. Rose, M. A. Behlke and A. K. Salem, Int. J. Pharm., 2012, 427, 123–133 CrossRef CAS PubMed.
- J. M. Li, Y. Y. Wang, W. Zhang, H. Su, L. N. Ji and Z. W. Mao, Int. J. Nanomed., 2013, 8, 2101–2117 CrossRef PubMed.
- M. Ogris, P. Steinlein, M. Kursa, K. Mechtler, R. Kircheis and E. Wagner, Gene Ther., 1998, 5, 1425–1433 CAS.
- M. Bauer, C. Lautenschlaeger, K. Kempe, L. Tauhardt, U. S. Schubert and D. Fischer, Macromol. Biosci., 2012, 12, 986–998 CrossRef CAS PubMed.
- K. Kunath, A. von Harpe, D. Fischer, H. Petersen, U. Bickel, K. Voigt and T. Kissel, J. Controlled Release, 2003, 89, 113–125 CrossRef CAS PubMed.
- D. Fischer, T. Bieber, Y. Li, H. P. Elsasser and T. Kissel, Pharm. Res., 1999, 16, 1273–1279 CrossRef CAS.
- A. C. Grayson, A. M. Doody and D. Putnam, Pharm. Res., 2006, 23, 1868–1876 CrossRef PubMed.
- S. K. Tripathi, R. Goyal, M. P. Kashyap, A. B. Pant, W. Haq, P. Kumar and K. C. Gupta, Biomaterials, 2012, 33, 4204–4219 CrossRef CAS PubMed.
- R. Goyal, S. K. Tripathi, S. Tyagi, A. Sharma, K. R. Ram, D. K. Chowdhuri, Y. Shukla, P. Kumar and K. C. Gupta, Nanomedicine, 2012, 8, 167–175 CAS.
- Y. Ren, X. Jiang, D. Pan and H. Q. Mao, Biomacromolecules, 2010, 11, 3432–3439 CrossRef CAS PubMed.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- R. Diaz-Lopez, N. Tsapis, M. Santin, S. L. Bridal, V. Nicolas, D. Jaillard, D. Libong, P. Chaminade, V. Marsaud, C. Vauthier and E. Fattal, Biomaterials, 2010, 31, 1723–1731 CrossRef CAS PubMed.
Footnotes |
| † Xuelan Tang, Ping Lu, and Mingfeng Qiu contributed equally to this work. |
| ‡ Permanent address: School of Pharmacy, Shanghai Jiao Tong University, 800, Dongchuan Road, Shanghai, 200240, China, Tel: +86-21-34204052. |
| This journal is © The Royal Society of Chemistry 2016 |