
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licencep-Bromoaryl- and ω-bromoalkyl-VA-PNBs: suitable starting materials for the functionalization of vinylic addition polynorbornenes via palladium-catalyzed cross-coupling reactions†
Sheila Martínez-Arranz,
Eduardo Sánchez-Pérez,
Jesús A. Molina de la Torre,
Ignacio Pérez-Ortega and
Ana C. Albéniz *
*
IU CINQUIMA/Química Inorgánica, Universidad de Valladolid, 47071 Valladolid, Spain. E-mail: albeniz@qi.uva.es
First published on 31st October 2016
Abstract
New vinylic addition polynorbornenes (VA-PNBs) with a variable amount of pendant 4-bromoaryl groups have been synthesized by homo- and copolymerization with norbornene of 2-(4-bromophenyl)-5-norbornene catalyzed by [Ni(C6F5)2(SbPh3)2]. These polymers can be transformed by Pd-catalyzed cross coupling reactions into VA-PNBs with pendant diaryl (Suzuki), alkenyl and ketoalkyl (Stille) and amino (Buchwald–Hartwig) groups. Although the cross-coupling reactions of alkylhalides are more difficult, (ω-bromobutyl)-VA-PNBs can also be functionalized by a Suzuki reaction with arylboronic acids. This results open up new ways of post-polymerization functionalization of VA-PNBs, quite attractive for their robust, thermally stable and transparent skeleton.
Introduction
Vinylic addition polynorbornenes (VA-PNBs, Fig. 1) have emerged as convenient and robust polymeric materials for a variety of applications. Their all-aliphatic polycyclic backbone makes them less reactive that their ring opening metathesis polymerization counterparts (ROMP-PNBs, Fig. 1),1 and it imparts good properties such as high thermal stability, high Tgs, and transparency. As a result, new applications of VA-PNBs are being explored to construct optical devices,2 ion exchange membranes,3 gas separation membranes,4 and supports in synthesis and catalysis,5 including photocatalysis.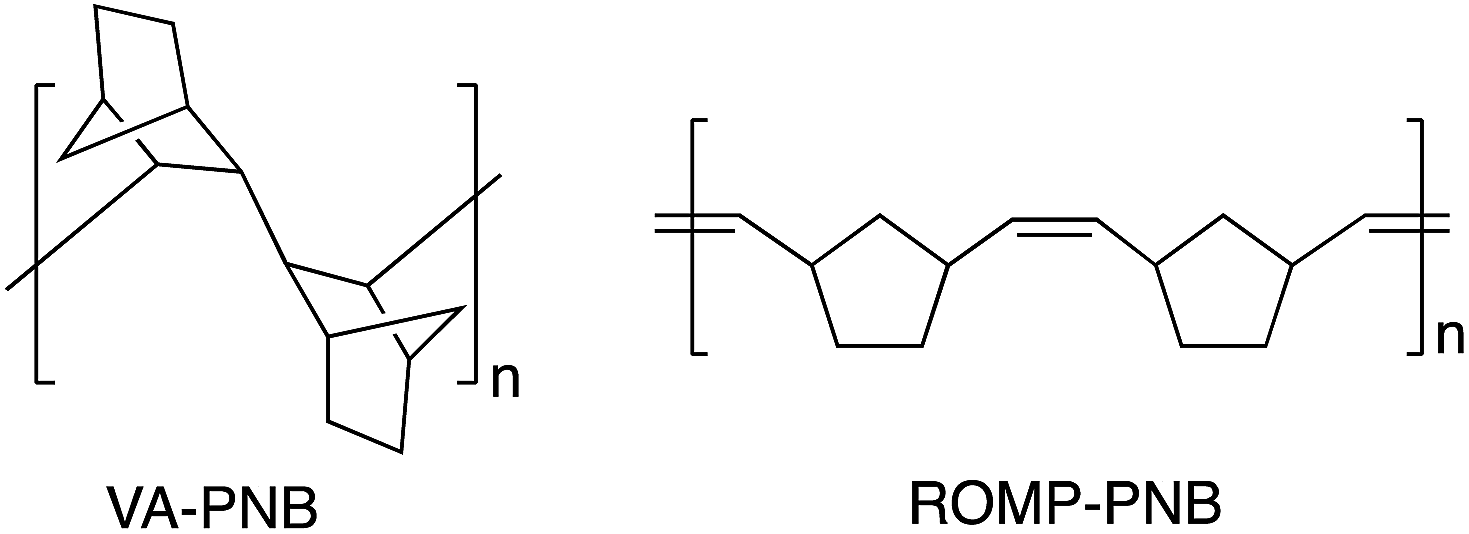 | ||
| Fig. 1 Types of polynorbornene backbone. | ||
Most applications require the presence of functional groups that either improve the properties of the polymeric material or carry the active site for the desired use (a luminescent group, a catalyst, etc.). Functionalized VA-PNBs are not easy to obtain by direct polymerization or copolymerization of substituted norbornenes. In contrast, the ROM-polymerization of norbornene is more tolerant to the presence of functional groups in the monomer, although it introduces double bonds on the ROMP-PNB skeleton (Fig. 1) that are potential centers for polymer degradation and can indeed react in the presence of metal complexes.1a If a hydrogenation reaction is applied, an all-aliphatic polymer backbone can be obtained,6 but many functional groups may not survive this process.7 The presence of polar groups notably decreases the reactivity of norbornenes towards double bond insertion polymerization (VA-polymerization) and most catalyst that are very active for the VA-polymerization of norbornene do not perform well with substituted norbornenes. Only a few nickel or palladium catalysts can polymerize substituted norbornenes (NB-R) or copolymerize them with norbornene to give VA-PNBs.2,3a,4,8,9 Additionally, since the NB-R are usually synthesized by a Diels–Alder reaction, they are a mixture of exo and endo isomers which have different polymerization rates and this is sometimes an extra difficulty.10 We reported a different strategy to synthesize functionalized VA-PNBs that circumvents this problem and involves the post-polymerization functionalization of a VA-PNB with pendant ω-bromoalkyl units.8 The bromoalkyl-polymer can be obtained in good yields and different bromo loadings by direct copolymerization of NB and NB-(CH2)nBr using [Ni(C6F5)2(SbPh3)2]. The nucleophilic substitution of bromide in the polymer leads to VA-PNBs with polar groups (ester, cyano, SR, imidazole, etc.),8,5b as well as the azido moiety that can be used as to anchor other groups by a click CuAAC reaction with alkynes.5c The versatility of these brominated VA-PNBs led us to explore the possibility of using Pd-catalyzed cross coupling reactions to functionalize VA-PNBs. For that, we envisioned that a VA-PNB with pendant bromoaryl groups would be a suitable parent material. Thus, we describe here the preparation of those polymers as well as the application of a variety of Pd-catalyzed cross coupling reactions to give VA-PNBs with pendant biaryls, anilines and other groups. The functionalization of the more challenging VA-PNBs with ω-bromoalkyl groups by the same methods is also reported.
Cross-coupling reactions catalyzed by palladium are extremely useful in synthesis and this has been highly recognized.11 In the realm of polymer chemistry these reactions have found application in the synthesis of polymeric materials by condensation of two doubly-functionalized coupling reagents. It is worth mentioning the Stille reaction for the preparation of a variety of polymers,12 the Suzuki and Sonogashira couplings in the synthesis of polyfluorenes,13 and some examples that involve the Heck reaction,14 the Buchwald–Hartwig amination,15 or the related ipso-arylation.16 Fewer examples can be found of the application of Pd-catalyzed C–C or C–X bond forming reactions for the post-polymerization functionalization of preformed polymers.17 Most examples report the use of the Sonogashira coupling to synthesize derivatives of polystyrene,18 or polyphenylene.19 Polytriazoles,20 and polythiophenes have also been subjected to this type of reactions.21 The functionalization of polymers by palladium-catalyzed C–N bond formation has been scarcely used on polystyrene,22 or PEG.23
Results and discussion
Synthesis of p-bromoaryl VA-PNBs
Polymers with pendant bromoaryl groups were synthesized by homopolymerization of 1 or co-polymerization of this monomer with norbornene (NB) (Scheme 1). Monomer 1 is not commercial and it was synthesized by a Diels–Alder reaction of 1-bromo-4-vinylbenzene with cyclopentadiene. In order to avoid radical side reactions 2,6-ditertbutylphenol was used as a trap. 1 is a mixture of isomers in a ratio endo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :exo = 80:20 and this mixture was used for the polymerization reactions.
:exo = 80:20 and this mixture was used for the polymerization reactions.
 | ||
| Scheme 1 Synthesis and polymerization of 1 to give p-bromophenyl-VA-PNBs. | ||
The polymerization reactions were carried out as shown in Scheme 1 and the results are collected in Table 1. [Ni(C6F5)2(SbPh3)2] was chosen as catalyst because of its high activity both in homopolymerization of norbornene and in the copolymerization of norbornene with substituted norbornenes with ω-haloalkyl or alkyltin groups.5d,f,8,24 It is also very easy to handle as it is stable to moisture and air. The concentration of 1 was the same in all the experiments and the amount of NB in the copolymerization reactions was varied to reach the required monomer feed ratio; a catalyst loading between 0.5 and 2 mol% was used depending on the experiment (it is referred to the total amount of monomer). Monomer 1 can be homopolymerized with this catalyst with a moderate yield to give polymer 2 (entry 1, Table 1). The copolymerization of NB and 1 gave polymers 3 with different composition depending on the monomer feed ratio (a/b = NB/1 ratio in the polymer, Scheme 1), and moderate to good yields, the better the higher the NB ratio in the feed mixture. All the polymers are obtained by precipitation in MeOH as off-white solids. The ratio a/b was determined by quantitative analysis of the Br content in the polymer.
| Entry | Feed ratiob NB:1:[Ni] |
Yieldc (%) | Polymer, a/bd | Mwe (Da) | Mw/Mne |
|---|---|---|---|---|---|
| a The reactions were carried out using CH2Cl2 as solvent, 0.4 mmol of 1 and a total volume of 9 mL (unless otherwise noted) at room temperature for 10 min.b Molar ratio in the feed.c Yields are referred to the total monomer mass.d a/b = NB/NBC6H4X molar ratio; it was determined by quantitative analyses of the bromine in the polymer.e Determined by GPC using polystyrene standards.f 0.80 mmol of 1 were used and a total volume of 18 mL; reaction time 24 h. | |||||
| 1f | 0:50:1 |
50 | 2, 0 | 8755 | 2.3 |
| 2 | 33:50:1 |
45 | 3, 1.3 | 37248 |
3.2 |
| 3 | 50:50:1 |
74 | 3, 1.8 | 49170 |
3.9 |
| 4 | 75:50:1 |
>99 | 3, 2.4 | 68521 |
3.8 |
| 5 | 100:50:1 |
>99 | 3, 4.7 | 101508 |
3.6 |
| 6 | 150:50:1 |
>99 | 3, 5.7 | 119405 |
2.4 |
The polymers synthesized showed unimodal distributions in GPC and a polydispersity in the range of 2.3–3.9. The molecular weight range was between 8 × 103 to 120 × 103 Da and it increased as the ratio of NB increased. When the ratio NB:1 in the feed was higher than those shown in Table 1, insoluble polymers were obtained that could not be characterized by GPC.
According to the data in Table 1 (entries 2–6), a rough determination of the reactivity ratios of NB and 1 can be made: rNB = 1.86, and r1 = 0.45.25 These values imply that NB has a higher probability than 1 of entering the copolymer, so the composition of copolymers 3 obtained do not reproduce the monomer feed ratio and are always richer in NB. A composition drift of the copolymer as the polymerization proceeds is also expected.
Polymers 2 and 3 were characterized by IR, 1H and 13C NMR. The NMR spectra in solution showed broad signals corresponding to the NB skeleton. The aromatic signals of the pendant bromoaryl groups were clearly observed in the 1H and 13C{1H} NMR spectra. No signals around 20 ppm were observed in the 13C NMR, which indicated exo-enchained norbornene units in the polymer backbone.26 Furthermore, in these polymers terminal pentafluorophenyl groups were observed in the 19F NMR spectra, which suggest an insertion polymerization mechanism with an initiation step that involves insertion of the olefin in the Ni–C6F5 bond.8,24,27 The IR spectra of polymers 2 and 3 clearly show a band at 1075 cm−1 due to the CAr–Br stretching. After obtaining the polymers, the analysis by 1H NMR of the mother liquors showed that in all the cases the unreacted endo-1:exo-1 ratio showed values in the range 100:0 to 91:9, higher than the endo-1:exo-1 = 80:20 ratio in the initial monomer. This clearly shows that, as expected, the exo isomer of 1 reacts preferently to the endo isomer in transition metal NB-insertion polymerization.
Functionalization of p-bromoaryl VA-PNBs
A simplified mechanism for palladium-catalyzed cross coupling reactions involves three main steps: oxidative addition of the organic halide, coordination of the nucleophilic reagent, by transmetalation in most cases, and reductive elimination (Scheme 2).28 The actual mechanism in each case may involve additional isomerizations, ligand coordination and exchange reactions, which are also important, but the representation in Scheme 2 can be used as a reasonable working guide. VA-PNBs 2 and 3, the organic halide reagents, should undergo oxidative addition to palladium, the first step in Scheme 2, if they are to be functionalized by these reactions. In order to test this point the reaction of 3 and [Pd(PPh3)4] was carried out (eqn (1)). Indeed the reaction occurred and a palladium-functionalized polymer, 4, was obtained. IR analysis showed the disappearance of the ν(CAr–Br) at 1075 cm−1 band and the appearance of the bands corresponding to the ν(Pd–P) at 539 cm−1. The formation of 4 also becomes clear when we recorded 13C CP-MAS and 31P CP-MAS spectra, where a signal at 26 ppm typical of a coordinated PPh3 to palladium in a trans “PdP2” arrangement, is observed.
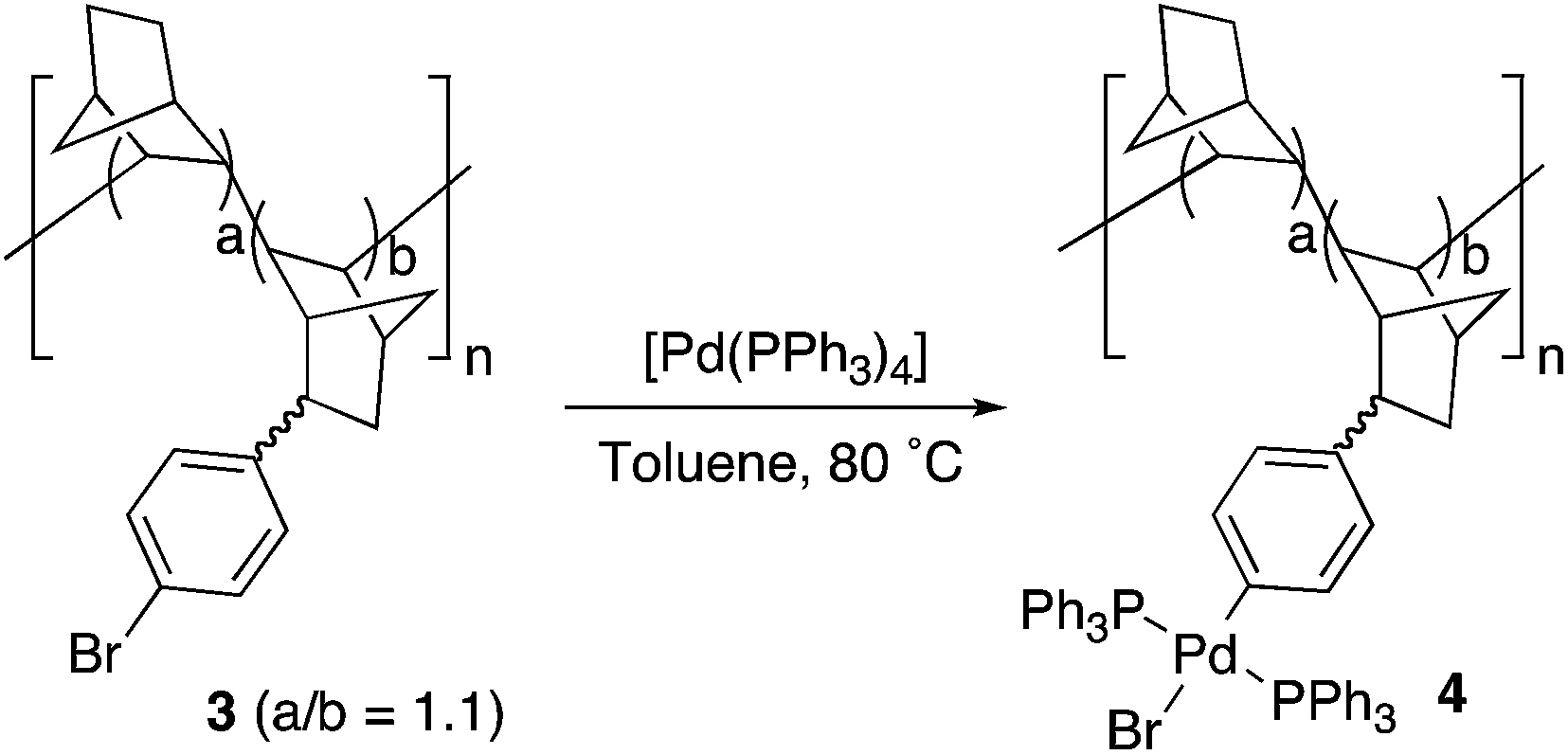 | (1) |
 | ||
| Scheme 2 Simplified catalytic cycle for Pd-catalyzed cross-coupling reactions. | ||
Once we found out that polymer 3 is a suitable reagent for the coupling processes, we tested the utility of the Suzuki reaction to introduce aryl groups on 3. Scheme 3 (top) collects the aryl groups introduced and Table 2 the reactions tried. Polymers 5–8 were obtained in good yields (82–98%) as greyish solids insoluble in common organic solvents.‡ Thus, they were characterized by IR and solid state MAS-NMR, and the most significant absorptions of each group are clearly visible with both techniques (see the Experimental section and ESI†). The extent of the reaction was measured by quantitative analyses of the residual bromine in the polymer products, and it is given as the percentage of Br that has been substituted in Table 2. Two different sets of reaction conditions were employed. Method A (entries 1–6, Table 2) uses the best reaction conditions we found using palladium catalysts with conventional aryl phosphines (see ESI† for additional optimization experiments). Using [PdCl2(dppf)] as precatalyst in toluene at 110 °C good results were obtained for the boronic acids BAr(OH)2 where Ar = Ph, 4-CF3–C6H4 (entries 1–4, Table 2). The reaction works well regardless the composition of the starting polymer 3, so polymers with different degree of functionalization can be obtained (entries 2–4, Table 2). Method A is less efficient for Ar = 4-OMe–C6H4, 2-Me–4OMe–C6H3 (62% of Br substituted, entries 5, 6, Table 2). However, the reaction conditions of Method B, used by Fu et al. for more challenging alkyl halide substrates, proved to be superior (see below). In the presence of the precatalyst system [Pd2(dba)3]/PtBu2Me and 2-methyl-2-butanol as solvent, almost complete reaction of the arylbromo substituents of the polymer was achieved (entries 7, 8, Table 2).
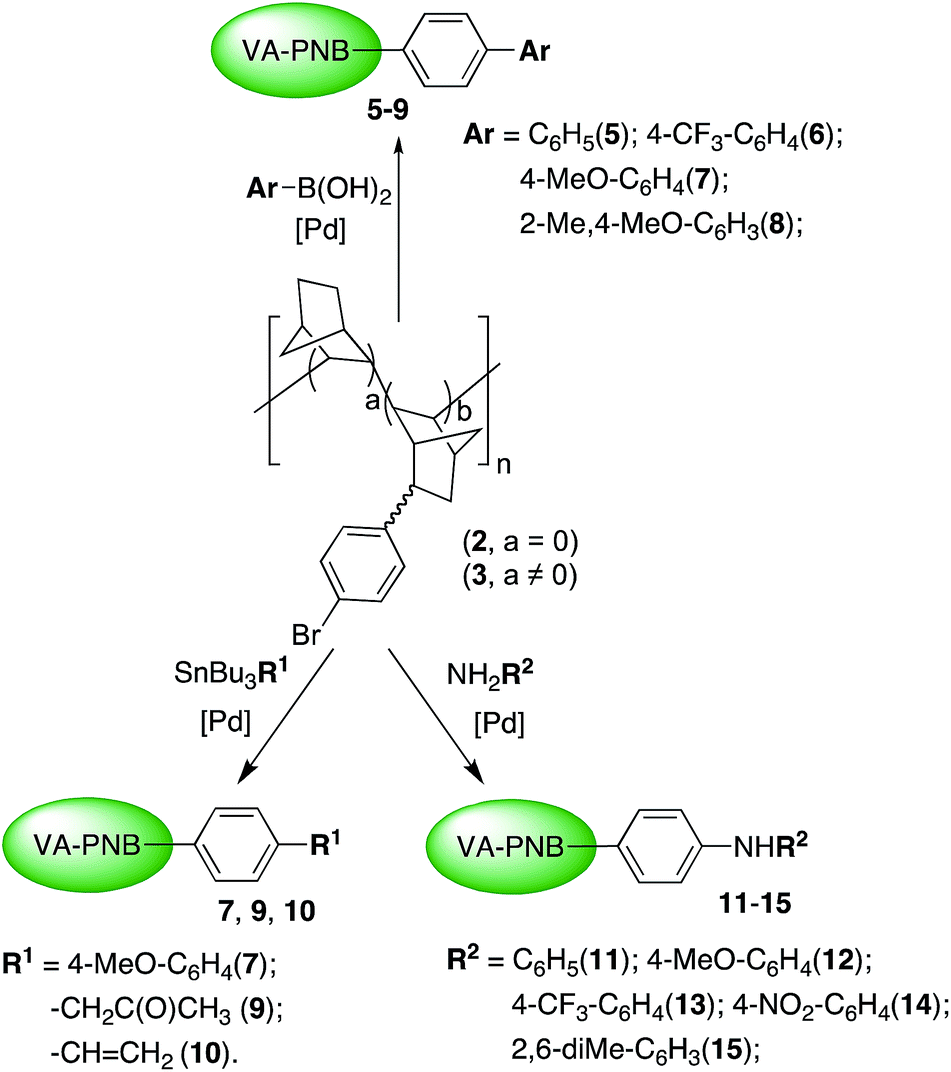 | ||
| Scheme 3 Functionalization of VA-PNBs 2 and 3 by Pd-catalyzed C–C and C–N coupling. | ||
| Entry | VA-PNB, a/b (mmol Br per g) | Method | Product VA-PNB | Ar | % Br subst.a |
|---|---|---|---|---|---|
| a Determined by quantitative analyses of the bromine content in the polymer product.b Method A: the reactions were carried out at 110 °C using toluene as solvent and [PdCl2dppf] as catalyst (1 mol%). Ratio of reagents: ArB(OH)2:VAPNB-C6H4-Br:K2CO3 = 2:1:2.5. Reaction time 48 h.c Method B: the reactions were carried out at 100 °C using t-amylalcohol as solvent, [Pd2dba3] as precatalyst (2.5 mol%), and PtBu2Me (10 mol%). Ratio of reagents: ArB(OH)2:VAPNB-C6H4-Br:KOtBu = 1.5:1:3. |
|||||
| 1 | 3, 1.6 (2.5) | Ab | 5 | C6H5 | 94 |
| 2 | 3, 1.3 (2.7) | Ab | 6 | 4-CF3–C6H4 | 94 |
| 3 | 3, 4 (1.6) | Ab | 6 | 4-CF3–C6H4 | 93 |
| 4 | 2, 0 (4) | Ab | 6 | 4-CF3–C6H4 | 89 |
| 5 | 3, 1.3 (2.7) | Ab | 7 | 4-MeO–C6H4– | 62 |
| 6 | 3, 1.6 (2.5) | Ab | 8 | 2-Me–4-MeO–C6H3– | 62 |
| 7 | 3, 1 (2.9) | Bc | 7 | 4-MeO–C6H4– | 95 |
| 8 | 3, 1.4 (2.6) | Bc | 8 | 2-Me–4-MeO–C6H3– | 98 |
The Suzuki reaction is the most convenient one for the formation of a pendant biaryl moiety on the polymer. The Stille reaction is a viable but less efficient alternative for this type of groups as shown in entry 3, Table 3. However the use of SnBu3R (R ≠ aryl) leads to VA-PNBs functionalized with ketoalkyl (9) or vinyl (10) groups in a very good or moderate Br substitution degree respectively (entries 1 and 2, Table 3). In the case of 9, the required organostannane can be easily prepared in situ from SnBu3OMe and 2-propenylacetate. Both 9 and 10 are insoluble in common solvents and, again, their characterization was made by IR and solid state MAS-NMR (see Experimental section).
| Entry | VA-PNB, a/b (mmol Br per g) | Product VA-PNB | R1 | % Br subst.b |
|---|---|---|---|---|
| a The reactions were carried out at 110 °C for 48 h using toluene as solvent.b Determined by quantitative analyses of the bromine content in the polymer product.c [PdCl2{P(o-MeC6H4)3}2] as catalyst (1 mol%). Ratio of reagents: SnBu3R1:VAPNB-C6H4-Br = 1.5:1.d [Pd(PPh3)4] as catalyst (10 mol%). Ratio of reagents: SnBu3R1:VAPNB-C6H4-Br = 1.1:1.e [Pd(PPh3)4] as catalyst (2 mol%). |
||||
| 1c | 3, 1.6 (2.5) | 9 | –CH2COCH3 | 98 |
| 2d | 3, 1 (2.9) | 10 | –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
75 |
| 3e | 3, 1.6 (2.5) | 7 | MeOC6H4– | 56 |
The Buchwald–Hartwig amination reaction is a good route for the formation of supported diaryl amines on VA-PNBs. The reaction of 3 in the presence of different anilines, a base and a mixture of [Pd2dba3] and X-Phos as precatalyst led to polymers 11–15 with complete bromo-substitution (Scheme 3 and Table 4). The reaction conditions followed those reported by Marqués et al. for the synthesis of PEG derivatives with pendant anilines.23 Amines with either electron-donating or electron-withdrawing groups on the aryl ring can be introduced (entries 1–4, Table 4). An increase in the steric bulk of the aniline is not a problem either (entry 5, Table 4). The IR spectra of polymers 11–15 clearly shows the disappearance of the ν(CAr–Br) at 1075 cm−l and a new weak resonance at about 3400 cm−l typical of the N–H stretching.
| Entry | VA-PNB, a/b (mmol Br per g) | Product VA-PNB | NHR2 | % Br subst.b |
|---|---|---|---|---|
| a The reactions were carried out at 110 °C for 48 h using toluene as solvent, [Pd2dba3] as precatalyst (1 mol%) and X-Phos (6 mol%). Ratio of reagents: R2-NH2:VAPNB-C6H4-Br:KOtBu = 2:1:2.b The polymer products do not contain any bromine as determined by quantitative analyses. |
||||
| 1 | 3, 1.4 (2.6) | 12 | NHC6H5 | >99 |
| 2 | 3, 1.4 (2.6) | 13 | NH–C6H4–4-MeO | >99 |
| 3 | 3, 1.3 (2.7) | 14 | NH–C6H4–3-CF3 | >99 |
| 4 | 3, 1.4 (2.6) | 15 | NH–C6H4–4-NO2 | >99 |
| 5 | 3, 1.4 (2.6) | 16 | NH–C6H3–2,6-Me2 | >99 |
Functionalization of ω-bromobutyl VA-PNB
A more challenging reaction is the functionalization of bromoalkyl VA-PNBs by Pd-catalyzed C–C cross coupling reactions. After oxidative addition of an alkylhalide (R1 = alkyl, Scheme 2), the transmetalation step competes with a favorable β-H elimination that often hampers the coupling process. Nonetheless, useful catalysts and reaction conditions have been developed, specially by Fu's group, that allow the use of alkylhalides (and also alkyl organometallics) as coupling partners.29 No Pd-catalyzed alkyl coupling has been tried on polymeric substrates so far, and we decided to test our ω-bromoalkyl-VA-PNBs reported before.8 Polymer 16 was synthesized by copolymerization of norbornene and 2-(ω-bromobutyl)-5-norbornene with [Ni(C6F5)2(SbPh3)2] in an analogous reaction to that in Scheme 1 and following the procedure we described in the literature.8The Suzuki reaction was tested on polymer 16 (2.7 mmol Br per g; a/b = 1.5) using the conditions described by Fu et al. for discrete alkylbromides (same as Method B in Table 2). Polymers 17–19 were obtained as greyish solids insoluble in most common organic solvents (Scheme 4). The substitution of bromide in the starting polymer 16 is almost complete in all cases (95–99%). This is shown by the quantitative determination of the bromide content in polymers 17–19 as well as the disappearance of the band at ν(CAlk–Br) at 562 cm−l present in 16.
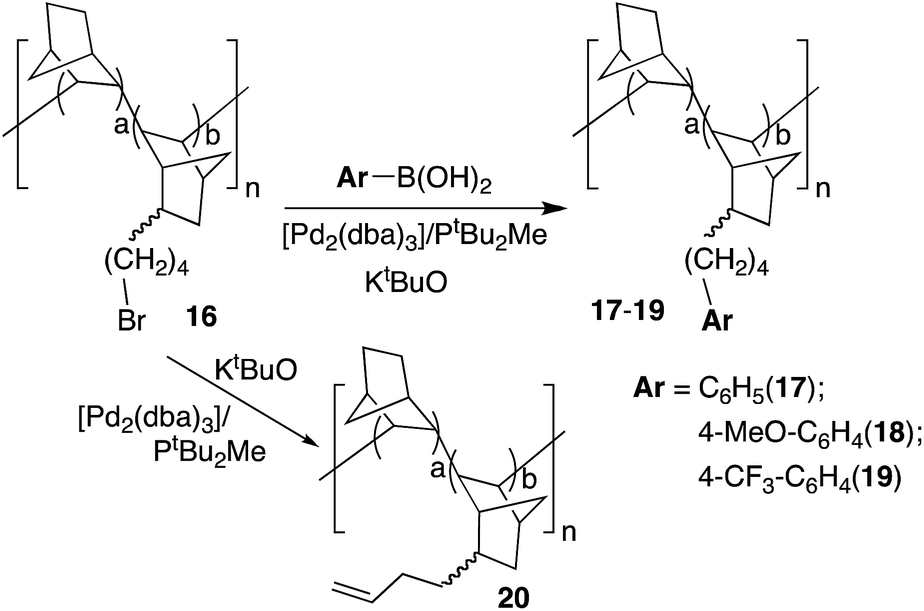 | ||
| Scheme 4 Functionalization of VA-PNB 16 by Suzuki coupling and Pd-catalyzed β-H elimination. | ||
Since the elimination of bromide could occur easily in this case by β-H elimination after oxidative addition of the ω-bromobutyl-VA-PNB, with little incorporation of the aryl moiety, we decided to check this possibility. Thus, we run the reaction in the same conditions used in the Suzuki couplings, but in the absence of the coupling partner, i.e. the boronic acid (Scheme 4). Polymer 20 was obtained which showed the presence of a terminal double bond both in the 13C MAS-NMR spectrum (signals at 140 and 116 ppm) and in the IR spectrum (absorptions at 1640 (m) and 908 (s) cm−1). The bromide substitution was complete. Fig. 2 shows the IR spectra of polymers 17 and 20. As can be seen, the β-H elimination is not important in the Suzuki reactions and the most intense bands in the IR of 17 correspond to the phenyl group incorporated (762, 696 cm−1). However, the presence of a terminal double bond cannot be completely excluded in 17 since there is a very low intensity band at 908 cm−1. The same applies to polymers 18 and 19.
 | ||
| Fig. 2 IR spectra of polymers 17 (a) and 20 (b). | ||
Elimination of the residual palladium in the functionalized polymers
We analyzed the polymer products after the functionalization reactions in order to find out how much residual palladium was trapped in them. The ICP-MS quantitative data showed that the amount of Pd varies from 1.5 mg g−1 in the reactions where 1% mol of palladium catalysts is used to 5–10 mg g−1 if the amount of catalyst in the reaction is 5% mol. Several procedures to eliminate this residual metal were tested. Since the palladium occluded in the polymer is most probably black Pd(0) coming from the decomposition of the catalyst once the coupling reagents have been consumed, we decided to check if an oxidative addition reaction, to give a soluble Pd(II) complex, was a suitable cleaning way. A sample of polymer 6 containing 1.19 mg Pd per g was mixed with an excess of 1-trifluoromethyl-4-iodobenzene and refluxed in toluene for 24 h. The analysis of the polymer showed that only about 10% of the residual Pd was eliminated (1 mg Pd per g remained). The reaction was carried out with a new sample of 6 (1.6 mg Pd per g) but adding PPh3 to increase the stability and solubility of the oxidative addition product (a putative [PdI(4-CF3–C6H4)(PPh3)2]). In this way half of the palladium in the polymer could be eliminated (from 1.6 mg Pd per g in 6 to 0.8 mg Pd per g after treatment).Krebs et al. have used N,N-dimethyl-2-phenyldiazene-carbothioamide to remove palladium nanoparticles from polymers synthesized by Pd-catalyzed reactions.30 They describe that this compound coordinates to Pd(0) and forms a soluble complex in most organic solvents. Thus, a sample of polymer 19 that contained 4.6 mg Pd per g was treated with an excess of N,N-dimethyl-2-phenyldiazenecarbothioamide and refluxed in toluene for 24 h. The cleaned 19 contained 0.47 mg Pd per g, one order of magnitude lower than the initial amount, indicating that this is a superior method to that tested above. In general, this treatment leads to polymers that contain a palladium residue ten times lower than the initial amount (see ESI†). The process can be repeated leading to a further reduction of Pd, although about 400 ppm is the lowest limit we have reached.
Experimental
Materials and general considerations
The compounds [Ni(C6F5)2(SbPh3)2],31 [PdCl2(dppf)],32 [Pd(PPh3)4],33 and [PdCl2(P(o-tolyl)3)2],34 [Pd2(dba)3],35 [PdCl2(PPh3)2],36 N,N-dietilfenilazotioformamida,30 tributyl(4-methoxyphenyl)-stannane,37 and tributyl(vinyl)stannane,37 were prepared according to the literature procedures. Polymer VA-PNBNB(CH2)4Br (16) was prepared as reported before.8 The other reagents used in this work were purchased from commercial suppliers and used without further purification. Solvents such as THF and CH2Cl2 were dried using a Solvent Purification System (SPS); toluene was dried over Na, distilled and deoxygenated prior to use. NMR spectra were recorded at 293 K using Bruker AV-400 and Agilent MR-500 instruments. Chemical shifts (δ) are reported in ppm and referenced to SiMe4 (1H, 13C) or CFCl3 (19F). The solid state NMR spectra were recorded at room temperature under magic angle spinning (MAS) in a Bruker AV-400 spectrometer using a Bruker BL-4 probe with 4 mm diameter zirconia rotors spinning at 10 kHz. 13C CP MAS NMR spectra were measured at 100.61 MHz and recorded with proton decoupling (tppm), with 90° pulse length of 4.5 μs and a contact time of 3 ms. Chemical shifts were calibrated indirectly through the glycine CO signal recorded at 176.0 ppm relative to TMS. 31P MAS NMR spectra were recorded at 161.97 MHz with proton decoupling (tppm), with a 90° pulse length of 5.45 μs. 31P-NMR chemical shifts are in ppm relative to external 85% H3PO4. 19F MAS NMR spectra were recorded at 376.5 MHz with a 90° pulse length of 5.5 μs. 19F NMR chemical shifts are in ppm relative to external CFCl3. IR spectra were recorded either on neat samples using a Perkin-Elmer FT/IR SPECTRUM FRONTIER spectrophotometer with CsI + ATR diamond accessory in the range 200–4000 cm−1, or on KBr pellets using a Perkin-Elmer BX FT-IR in the range 400–4000 cm−1. Size exclusion chromatography (SEC) was carried out using a Waters SEC system on a three-column bed (Styragel 7.8 × 300 mm columns: 50–105, 5 × 103 to 5 × 105 and 2 × 103 to 4 × 106 Da) and a Waters 410 differential refractometer. SEC samples were run in CHCl3 at 313 K and calibrated to polystyrene standards. The bromine content in the polymers was determined by oxygen-flask combustion of a sample and analysis of the residue by mercurimetric titration of the bromide.38 The bromine content of the polymer 3 (x = mmol Br per g) and the polymer composition given as a ratio of monomers incorporated (a/b = NB/1) are related by the equation: x = 1000/(94.16(a/b) + 249.155), where 94.16 and 249.155 are the molecular weights of norbornene and NB-C6H4Br (1) respectively. The amount of residual palladium in the functionalized polymers was determined by ICP-MS, using Agilent 7500i equipment; the samples were dissolved in HNO3 (65%) using an ETHOS SEL Milestone microwave oven. Each analysis is the average of two independent determinations for each sample. C, H and N elemental analyses were carried out using a Perkin-Elmer 2400 CHN microanalyzer.:20 endo:exo mixture of isomers (1.24 mL, 44% yield). Elemental analysis: calcd (%) for C13H13Br: C, 62.67; H, 5.26; found: C, 62.45; H, 5.20. MS (EI, m/z (%)): 250, 248 (10) [M+], 184, 182 (100) [M+ − cyclopentene], 103 (26) [M+ − cyclopentene − Br], 77 (24) [M+ − NB − Br], 66 (50) [M+ − CH2CHC6H4Br].
1exo. 1H NMR (400.13 MHz, CDCl3): δ 7.42 (m, 2H, H9, H13), 7.16 (m, 2H, H10, H 12), 6.26 (dd, J = 5.71, 2.78 Hz, 1H, H6), 6.18 (dd, J = 5.71, 2.94 Hz, 1H, H5), 2.97 (br, 1H, H4), 2.86 (br, 1H, H1), 2.68 (m, 1H, H2), 1.75–1.6 (m, 2H, H3), 1.5 (m, 2H, H7). 13C {1H} NMR (100.6 MHz, CDCl3): δ 145.20 (s, 1C, C11), 137.47 (s, 1C, C5), 137.13 (s, 1C, C6), 131.25 (s, 2C, C9, C13), 129.35 (s, 2C, C10, C12), 119.19 (s, 1C, C8), 48.12 (s, 1C, C1), 45.72 (s, 1C, C7), 43.29 (s, 1C, C2), 42.33 (s, 1C, C4), 33.77 (s, 1C, C3).
1endo. 1H NMR (400.13 MHz, CDCl3): δ 7.35 (m, 2H, H9, H13), 7.02 (m, 2H, H10, H12), 6.26 (dd, J = 5.71, 3.1 Hz, 1H, H5), 5.78 (dd, J = 5.71, 2.78 Hz, 1H, H6), 3.35 (m, J = 9.29, 4.78 Hz, 1H, H2), 3.07 (br, 1H, H1), 2.97 (br, 1H, H4), 2.2 (ddd, J = 11.9, 9.29, 3.75 Hz, 1H, H3), 1.5 (m, 2H, H7), 1.26 (ddd, J = 11.3, 4.89, 2.61 Hz, 1H, H3′). 13C {1H} NMR (100.6 MHz, CDCl3): δ 144.05 (s, 1C, C11), 137.41 (s, 1C, C5), 132.58 (s, 1C, C6), 130.78 (s, 2C, C9, C13), 129.84 (s, 2C, C10, C12), 119.34 (s, 1C, C8), 50.27 (s, 1C, C7), 48.60 (s, 1C, C1), 43.23 (s, 1C, C4), 43.05 (s, 1C, C2), 33.04 (s, 1C, C3).
:NBC6H4Br = a:b = 1.8:1 ratio. The other copolymerization reactions were carried out in a similar way using a different monomer feed ratio as collected in Table 1.Functionalization of VA-PNBNBC6H4Br
CH2 (0.082 mL, 0.7504 mmol) and [PdCl2(P(o-tolyl)3)2] (0.004 g, 0.005 mmol) in a N2 atmosphere, was heated at 110 °C for 48 h. After that time, the solution was poured onto MeOH (30 mL), then was filtered, washed several times with MeOH (3 × 5 mL) and dried under vacuum. The product was obtained as a greyish solid. Isolated yield: 0.1773 g (77%). The copolymer contains 0.07 mmol Br per g, which indicates 98% of Br substitution. 13C CP-MAS NMR (100.61 MHz): δ 205 (s, 1C, CO), 151–139 (br, 1C, C11), 138–121 (br, 5C, C8–C10, C12, C13), 67–17 (br, 9C, C1–C 7, CH2C(O)CH3). IR (cm−1); ν(CO): 1720 (s).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH2 (10). A mixture of polymer 3 (0.1 g, 0.2865 mmol of Br), SnBu3CHCH2 (0.095 mL, 0.3152 mmol), [Pd(PPh3)4] (0.0331 g, 0.0286 mmol) and dry toluene (10 mL) was heated in a Schlenk flask under a N2 atmosphere at 110 °C for 48 h. After that time, the mixture was poured onto MeOH (30 mL). The solid was filtered, washed several times with MeOH (3 × 5 mL) and air-dried. The product was obtained as a greyish solid. Isolated yield: 0.0917 g (99%). The copolymer contains 0.76 mmol Br per g, which indicates 75% of Br substitution. 13C CP-MAS NMR (100.61 MHz): δ 154–140 (br, 2C, C8, C11), 140–135 (br, 2C, CHCH2), 135–127 (br, 4C, C9, C10, C12, C13), 67–22 (br, 7C, C1–C7). IR (cm−1); ν(CCst): 1629 (w), ν(CHδoop): 902 (m).
CH2 (10). A mixture of polymer 3 (0.1 g, 0.2865 mmol of Br), SnBu3CHCH2 (0.095 mL, 0.3152 mmol), [Pd(PPh3)4] (0.0331 g, 0.0286 mmol) and dry toluene (10 mL) was heated in a Schlenk flask under a N2 atmosphere at 110 °C for 48 h. After that time, the mixture was poured onto MeOH (30 mL). The solid was filtered, washed several times with MeOH (3 × 5 mL) and air-dried. The product was obtained as a greyish solid. Isolated yield: 0.0917 g (99%). The copolymer contains 0.76 mmol Br per g, which indicates 75% of Br substitution. 13C CP-MAS NMR (100.61 MHz): δ 154–140 (br, 2C, C8, C11), 140–135 (br, 2C, CHCH2), 135–127 (br, 4C, C9, C10, C12, C13), 67–22 (br, 7C, C1–C7). IR (cm−1); ν(CCst): 1629 (w), ν(CHδoop): 902 (m).Polymers 19 and 20 were prepared in the same way but using the corresponding boronic acid.
CH2 (20). A mixture of polymer 17 (0.150 g, 0.4006 mmol), KtBuO (0.147 g, 1.470 mmol), P(tBu)2Me (0.0078 mL, 0.04 mmol), Pd2(dba)3 (0.0104 g, 0.01 mmol) and 2-methyl-2-butanol (10 mL) was placed in a Schlenk flask and stirred for 48 h at 100 °C, under a N2 atmosphere. After this time, the resulting mixture was poured onto MeOH (30 mL) and stirred for 30 minutes. The resulting solid was collected by vacuum filtration and washed with MeOH (5 × 5 mL) and air-dried. The polymer was obtained as a greyish solid. Isolated yield: 0.1037 g (88%). The copolymer contains 0.05 mmol Br per g, which indicates 99% of Br substitution. 13C CP-MAS NMR (100.61 MHz): δ 140 (s, 1C, CH = CH2), 116 (s, 1C, CHCH2), 65–20 (br, 9C, C1–C7, 2CH2). IR (cm−1): ν(CCst): 1640 (m), ν(CHδoop): 908 (s).Conclusions
Vinylic addition polynorbornenes (VA-PNBs) with pendant p-bromophenyl groups can be synthesized by homopolymerization of 1 and copolymerization of 1 and norbornene catalyzed by [Ni(C6F5)2(SbPh3)2]. They are most convenient starting materials for the synthesis of VA-PNBs with aryl, ketoalkyl, alkenyl, and amino functional groups by Pd-catalyzed cross coupling reactions, as has been demonstrated in the application of the Suzuki, Stille and Buchwald–Hartwig amination reactions. A successful aryl functionalization by a Suzuki coupling can be applied to the more challenging ω-bromoalkyl-VA-PNB polymer. Post-polymerization functionalization is a very attractive route to synthesize functional VA-PNBs, which are difficult to synthesize by direct polymerization of substituted norbornenes. This opens up new ways to anchor a suitable functionality to the robust and transparent scaffold that the VA-PNB skeleton provides and, thus, increase the range of its applications as a support.Acknowledgements
We would like to thank the financial support of the Spanish MINECO (SE I+D+i, grant CTQ2013-48406-P), the MEC (FPU fellowship to JAMT) and the Junta de Castilla y León (grant VA302U13).Notes and references
- (a) R. García-Loma and A. C. Albéniz, RSC Adv., 2015, 5, 70244–70254 RSC; (b) M. Eo, D. Han, M. H. Park, M. Hong, Y. Do, S. Yoo and M. H. Lee, Eur. Polym. J., 2014, 51, 37–44 CrossRef CAS.
- (a) J. H. Park, C. Yun, T.-W. Koh, Y. Do, S. Yoo and M. H. Lee, J. Mater. Chem., 2011, 21, 5422–5429 RSC; (b) J. H. Park, C. Yun, M. H. Park, Y. Do, S. Yoo and M. H. Lee, Macromolecules, 2009, 42, 6840–6843 CrossRef CAS.
- (a) X. He, J. Liu, H. Zhu, Y. Zheng and D. Chen, RSC Adv., 2015, 5, 63215–63225 RSC; (b) J.-C. Daigle, V. Dube-Savoie, A. C. Tavares and J. P. Claverie, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2669–2676 CrossRef CAS; (c) H. Yao, X. He, L. Chen, Y. Chen and D. J. Chen, Appl. Polym. Sci., 2013, 3540–3547 CrossRef CAS.
- (a) M. V. Bermeshev, A. V. Syromolotov, M. L. Gringolts, L. E. Starannikova, Y. P. Yampolskii and E. S. Finkelshtein, Macromolecules, 2011, 44, 6637–6640 CrossRef CAS; (b) M. Gringolts, M. Bermeshev, Y. Yampolskii, L. Starannikova, V. Shantarovich and E. Finkelshtein, Macromolecules, 2010, 43, 7165–7172 CrossRef CAS.
- (a) J. A. M. de la Torre and A. C. Albéniz, ChemCatChem, 2016, 8, 2241–2248 CrossRef; (b) J. A. M. de la Torre and A. C. Albéniz, ChemCatChem, 2014, 6, 3547–3552 CrossRef; (c) I. K. Sagamanova, S. Sayalero, S. Martínez-Arranz, A. C. Albéniz and M. A. Pericàs, Catal. Sci. Technol., 2015, 5, 754–764 RSC; (d) N. Carrera, E. Gutiérrez, R. Benavente, M. M. Villavieja, A. C. Albéniz and P. Espinet, Chem.–Eur. J., 2008, 14, 10141–10148 CrossRef CAS PubMed; (e) I. Meana, A. C. Albéniz and P. Espinet, Adv. Synth. Catal., 2010, 352, 2887–2891 CrossRef CAS; (f) S. Martínez-Arranz, N. Carrera, A. C. Albéniz, P. Espinet and A. Vidal-Moya, Adv. Synth. Catal., 2012, 354, 3551–3560 CrossRef.
- Polymers based on the ROMP-hydrogenated polynorbornene are commercialized under the trademark Zeonex® by ZEON corporation. (a) M. Yamazaki, J. Mol. Catal. A: Chem., 2004, 213, 81 CrossRef CAS; (b) J. C. Mol, J. Mol. Catal. A: Chem., 2004, 213, 39 CrossRef CAS.
- We have developed ROMP-PNBs with haloalkyl groups that can be hydrogenated and lead to robust polymers that can be used, for example, as starting materials for the support of reagents in the Stille reaction. Their performance is comparable to the analogous VA-PNBs, although they need an additional synthetic step for their preparation (see ref. 1a).
- S. Martínez-Arranz, A. C. Albéniz and P. Espinet, Macromolecules, 2010, 18, 7482–7487 CrossRef and references therein.
- (a) F. Pierre, B. Commarieu, A. C. Tavares and J. Claverie, Polymer, 2016, 86, 91–97 CrossRef CAS; (b) B. Commarieu, J. Potier, M. Compaore, S. Dessureault, B. L. Goodall, X. Li and J. Claverie, Macromolecules, 2016, 49, 920–925 CrossRef CAS; (c) P. Hou, W. Liu, X. He, H. Wang and Y. Chen, Organometallics, 2013, 32, 2291–2299 CrossRef; (d) S. V. Mulpuri, J. Shin, B.-G. Shin, A. Greiner and D. Y. Yoon, Polymer, 2011, 52, 4377–4386 CrossRef CAS; (e) K. Müller, Y. Jung, D. Y. Yoon, S. Agarwal and A. Greiner, Macromol. Chem. Phys., 2010, 211, 1595–1601 CrossRef.
- (a) B. Commarieu and J. P. Claverie, Chem. Sci., 2015, 6, 2172–2181 RSC; (b) J. K. Funk, C. E. Andes and A. Sen, Organometallics, 2004, 23, 1680–1683 CrossRef CAS.
- (a) Nobel Foundation: http://nobelprize.org/nobel_prizes/chemistry/laureates/2010/press.html; (b) X.-F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9047–9050 CrossRef CAS PubMed.
- (a) M. Kaur, D. S. Yang, J. Shin, T. W. Lee, K. Choi, M. J. Cho and D. H. Choi, Chem. Commun., 2013, 49, 5495–5497 RSC; (b) B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed.
- (a) R. Molina, S. Gómez-Ruiz, F. Montilla, A. Salinas-Castillo, S. Fernández-Arroyo, M. M. Ramos and R. Mallavia, Macromolecules, 2009, 42, 5471–5477 CrossRef CAS; (b) Y. Tezuka, R. Komiya, Y. Ido and K. Adachi, React. Funct. Polym., 2007, 67, 1233–1242 CrossRef CAS; (c) C. Xue, V. R. R. Donuru and H. Liu, Macromolecules, 2006, 39, 5747–5752 CrossRef CAS; (d) R. Mallavia, F. Montilla, I. Pastor, P. Velásquez, B. Arredondo, A. L. Alvarez and C. R. Mateo, Macromolecules, 2005, 38, 3185–3192 CrossRef CAS; (e) F. Huang, H. B. Wu, D. Wang, W. Yang and Y. Cao, Chem. Mat., 2004, 16, 708–716 CrossRef CAS; (f) U. H. F. Bunz, Chem. Rev., 2000, 100, 1605–1644 CrossRef CAS PubMed.
- M. Winkler, L. Montero de Espinosa, C. Barner-Kowollik and M. A. R. Meier, Chem. Sci., 2012, 3, 2607–2615 RSC.
- T. Kanbara, K. Izumi, T. Narise and K. Hasegawa, Polym. J., 1998, 30, 66–68 CrossRef CAS.
- Y. S. Park, Q. Wu, C.-Y. Nam and R. B. Grubbs, Angew. Chem., Int. Ed., 2014, 53, 10691–10695 CrossRef CAS PubMed.
- Pd-catalyzed cross coupling reactions are carried out on polymeric solid supports to synthesize small molecules of interest, followed by their cleavage from the support after the coupling process (solid-phase synthesis). This is a widespread use of these reactions on polymers, but the goal of the process is not the synthesis of a new polymeric material. See for example: S. Bräse, J. H. Kirchhoff and J. Köbberling, Tetrahedron, 2003, 59, 885–939 CrossRef.
- (a) C. Kub, J. Tolosa, A. J. Zucchero, P. L. McGrier, C. Subramani, A. Khorasani, V. M. Rotello and U. H. F. Bunz, Macromolecules, 2010, 43, 2124–2129 CrossRef CAS; (b) L. B. Sessions, B. R. Cohen and R. B. Grubbs, Macromolecules, 2007, 40, 1926–1933 CrossRef CAS; (c) A. B. Morgan and J. M. Tour, Macromolecules, 1998, 31, 2857–2865 CrossRef CAS.
- (a) F. Bertini, G. Audisio, J. Kiji and M. Fujita, J. Anal. Appl. Pyrolysis, 2003, 68–69, 61–81 CrossRef CAS; (b) E. B. Stephens and J. M. Tour, Macromolecules, 1993, 26, 2420–2427 CrossRef CAS.
- E. Schwartz, K. Breitenkamp and V. V. Fokin, Macromolecules, 2011, 44, 4735–4741 CrossRef CAS.
- Y. Li, G. Vamvounis, J. Yu and S. Holdcroft, Macromolecules, 2001, 34, 3130–3132 CrossRef CAS.
- (a) T. Kanbara, Y. Yokokawa and K. Hasegawa, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 28–34 CrossRef CAS; (b) T. Kanbara, M. Oshima and K. Hasegawa, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 2155–2160 CrossRef CAS.
- L. Carvalho, M. Dias Pires, E. Fernandes and M. Marques, RSC Adv., 2013, 3, 25711–25715 RSC.
- (a) J. A. Casares, P. Espinet, J. M. Martín-Alvarez, J. M. Martínez-Ilarduya and G. Salas, Eur. J. Inorg. Chem., 2005, 3825–3831 CrossRef CAS.
- The composition data of the copolymers (F) obtained for the copolymerization at different monomer feed ratios (f) were fitted to the Finemann–Ross equation: J. M. G. Cowie, Polymers: Chemistry & Physics of Modern Materials, Chapman & Hall, Cheltenham, 1991 Search PubMed.
- (a) W. Kaminsky, A. Bark and M. Arndt, Makromol. Chem., Macromol. Symp., 1991, 41, 83–93 CrossRef; (b) M. Arndt, R. Engehausen, W. Kaminsky and K. Zoumis, J. Mol. Catal. A: Chem., 1995, 101, 171–178 CrossRef CAS.
- (a) D. A. Barnes, G. M. Benedikt, B. L. Goodall, S. S. Huang, H. A. Kalamarides, S. Lenhard, L. H. McIntosh, K. T. Selvy, R. A. Shick and L. F. Rhodes, Macromolecules, 2003, 36, 2623–2632 CrossRef CAS.
- Some references with mechanistic information: (a) Metal Catalyzed Cross-Coupling Reactions, ed. A. Meijere and F. Diederich, Wiley-VCH, Weinheim, 2004 Search PubMed. Stille reaction: (b) J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef; (c) C. Cordovilla and C. Bartolomé, ACS Catal., 2015, 5, 3040–3053 CrossRef CAS. Suzuki reaction: (d) C. Amatore, G. Le Duc and A. Jutand, Chem.–Eur. J., 2013, 19, 10082–10093 CrossRef CAS PubMed. Amination: (e) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046–2067 CrossRef CAS.
- (a) Z. Lu and G. C. Fu, Angew. Chem., Int. Ed., 2010, 49, 6676–6678 CrossRef CAS PubMed; (b) J. H. Kirchhoff, M. R. Netherton, I. D. Hills and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 13662–13663 CrossRef CAS PubMed; (c) J. H. Kirchhoff, C. Dai and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 1945–1947 CrossRef CAS; (d) M. R. Netherton, C. Dai, K. Neuschütz and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 10099–10100 CrossRef CAS PubMed.
- K. T. Nielsen, K. Bechgaard and C. Krebs, Macromolecules, 2005, 38, 658–659 CrossRef CAS.
- (a) A. Arcas and P. Royo, Inorg. Chim. Acta, 1978, 30, 205–207 CrossRef CAS; (b) G. Lopez, G. García, G. Sánchez, J. García, J. Ruiz, J. A. Hermoso, A. Vegas and M. Martínez-Ripoll, Inorg. Chem., 1992, 31, 1518–1523 CrossRef CAS.
- T. Hayashi, M. Konishi, Y. Kobori, K. Hirotsuf, M. Kumada and T. Higuchi, J. Am. Chem. Soc., 1984, 106, 158–163 CrossRef CAS.
- D. R. Coulson, L. C. Satek and S. O. Grim, Inorg. Synth., 1990, 28, 107–109 CrossRef CAS.
- J. Chatt and F. G. Mann, J. Chem. Soc., 1939, 1622–1634 RSC.
- T. Ukai, H. Kawazura, Y. Ishii, J. J. Bonnet and J. A. Ibers, J. Organomet. Chem., 1974, 65, 253–266 CrossRef CAS.
- H. A. Tayim, A. Bouldoukian and F. J. Awad, J. Inorg. Nucl. Chem., 1970, 32, 3799–3803 CrossRef CAS.
- A. L. Casado and P. Espinet, J. Am. Chem. Soc., 1998, 120, 8978–8985 CrossRef CAS.
- D. C. White, Microchim. Acta, 1961, 49, 449–456 CrossRef.
- Norbornene is a waxy solid, which sublimes easily. In order to accurately measure small quantities of this monomer, we used solutions of NB in CH2Cl2, previously titrated by 1H NMR.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional tables with experimental data, and IR and solution or solid state NMR spectra of the polymers. See DOI: 10.1039/c6ra23123c |
| ‡ After functionalization, all the polymers synthesized in this work are not soluble in most common organic solvents. We could not determine the exact reason of this change in properties. There is no loss of bromine when the homopolymer 2 was subjected to prolonged heating (48 h) in toluene or 2-methyl-2-butanol at T = 100–110 °C, indicating that no cross-linking derived from unwanted reactions of this group alone takes place. It has been shown that the rigid nature of the VA-PNB skeleton leads to polymers in metastable conformations that can rearrange upon heating and produce a change of properties (see ref. 5f and P. P. Chu, W.-J. Huang, F.-C. Chang, S. Y. Fan, Polymer 2000, 41, 401–404). This could have some importance along with the introduction of an additional rigid aromatic group that alters the morphology of the material and consequently its response to solvation. The occurrence of a small amount of homocoupling in the bromoaryl derivatives 3 (less likely in the bromoalkyl PNBs 16) may also contribute to the change in solubility and cannot be ruled out. |
| This journal is © The Royal Society of Chemistry 2016 |