
Preparation, characterization and oxygen reduction catalytic activities of nanocomposites of Co(II)/montmorillonite containing polypyrrole, polyaniline or poly(ethylenedioxythiophene)†
K. G. C. Senarathna,
H. M. S. P. Randiligama and
R. M. G. Rajapakse*
Department of Chemistry, Post-graduate Institute of Science, University of Peradeniya, Peradeniya 20400, Sri Lanka. E-mail: kgcsenarathna@gmail.com
First published on 23rd November 2016
Abstract
Electronically conducting composites of cobalt(II)/conducting polymer/montmorillonite are synthesized through ion exchange of montmorillonite (MMT) followed by oxidative polymerization of the monomers such as aniline, pyrrole or ethylenedioxythiophene within the interlayer spaces of the clay particles using Co(III) as the oxidant. In these syntheses, bentonite clay is purified to obtain pure MMT containing Na+ interlayer cations. Then, cobalt(III) ions are exchanged for Na+ ions present within the interlayer spaces. Monomers (aniline, pyrrole or 3,4-ethylenedioxythiophene) are then added to the ion-exchanged MMT dispersions. Co(III) is then reduced to Co(II) while oxidatively polymerizing aniline, pyrrole or 3,4-ethylenedioxythiophene monomers to polyaniline (PANI), polypyrrole (PPY) and poly(3,4-ethylenedioxythiphene) (PEDOT) respectively, thus forming Co(II)/MMT/PANI, Co(II)/MMT/PPY and Co(II)/MMT/PEDOT composite materials. These composites are characterized using XRD, XRF, FTIR, XPS, SEM and TGA analyses. These analyses show that in Co(II)/MMT/PANI and Co(II)/MMT/PPY composites the respective polymer and Co(II) ions are intercalated within the interlayer spaces of MMT while Co(II)/MMT/PEDOT shows exfoliated MMT platelets in the polymer/Co(II) matrix. The materials are also characterized for their electrical conductivities through DC conductivity measurements and AC impedance analyses and Co(II)/MMT/PANI, Co(II)/MMT/PPY and Co(II)/MMT/PEDOT have conductivities of 0.38 S m−1, 0.78 S m−1 and 0.32 S m−1 respectively. Electrochemical data depict that all three composites are good catalysts for the oxygen reduction half-reaction, and particularly Co(II)/MMT/PPY and Co(II)/MMT/PEDOT show good catalytic properties comparable to those of conventional and expensive C/Pt catalysts that are currently used in fuel cells. As such, very low-cost oxygen reduction catalysts for fuel cell applications can be realized with these clay/polymer/Co(II) systems.
Introduction
A fuel cell (FC) is an electrochemical device that can convert the chemical energy of the reaction between a fuel and an oxidant (−ΔG) into electrical energy (nFEcell). A fuel oxidizes at the anode and an oxidant reduces at the cathode and the anode and the cathode are separated by an electrolyte that permits the mass-transfer of ions between the two electrodes. In FCs, reactants are supplied externally in a continuous manner to maintain constant concentrations in order to consistently generate electricity and products are also discarded continuously. The most popular FCs utilize molecular hydrogen as the fuel and the common oxidant is the molecular oxygen and their reaction produce only water as the product. Hence, this kind of FC system is 100% green and renewable, which in theory, can provide solutions to future energy crisis and global warming. Moreover, FC is the highest efficient way to generate electricity from stored fuels when compared to gasoline, electric, diesel turbines, steam and gas turbines, coal heat turbines etc. Important properties of different FCs are collected in ESI Table S1.†The theoretical maximum electrical energy output, We, under standard conditions, in an FC is equal to the negative value of the Gibbs energy, ΔG of the reaction. In a typical H2/O2 FC, the following reaction takes place and the reaction has standard Gibbs energy (ΔGo) of −237 kJ mol−1 and standard enthalpy (ΔHo) of −286 kJ mol−1.
| ½O2 (g) + H2 (g) → H2O (l) | (1) |
Hence, in theory, 237 kJ of energy can be produced from one mole (or ∼2.0 g) of hydrogen gas, though the actual value depends on the efficiency of the cell. The maximum theoretical efficiency of a fuel cell (ΔGo/ΔHo) is 83% (under standard conditions) but, in practice, this is less than the theoretical value and their mean values are depicted in the ESI Table S1.†
Although, reaction (1) is thermodynamically spontaneous, it is not kinetically favorable. That is why the mixture of hydrogen and oxygen gases at ambient conditions (25 °C, 1 atm) is stable and the spontaneous combustion does not take place unless the required activation energy is supplied or the activation energy is brought down by using a suitable catalyst. The anodic half-reaction for hydrogen/oxygen FC is the oxidation H2 (g) which has standard electrode potential of 0.00 V with respect to (wrt) Standard Hydrogen Electrode (SHE) and the common cathodic half-reaction is the oxygen reduction half-reaction (ORR). There are two possible ways for ORR, depending upon the number of electrons transferred per reaction event (4e or 2e) and acidic/basic nature of the medium. Necessary background of thermodynamic and kinetic aspects of ORR is reviewed in ESI S1.†
Turning to the topic of ORR catalysts, the ideal catalyst for ORR should have following properties: high catalytic activity towards ORR, high electrical conductivity, high chemical and electrochemical stability, insolubility in the electrolyte (should be insoluble in acidic or basic aqueous solutions or in methanolic solutions), favourable optimum structural composition, favourable morphology, high specific surface area (SSA), small particle size, high porosity, uniform distribution of catalyst particles on the support, high interactions between the catalyst particle and the support surface, high catalytic stability and regeneration ability.1 Transition metals and metal oxides are normally used as catalysts in many purposes. However, the most of them have not shown good catalytic performance towards ORR except a few noble metals. The Sabatier principle is a qualitative concept in chemical catalysis and according to the principle, the interactions between the catalyst and the substrate should not be too strong or too weak because too weak interactions lead to failure of making bonds between the catalyst and the substrate, hence no reaction will take place, and too strong interactions lead to slow dissociation of the substrate/intermediates or products and then the catalytic surface will not be available for further reaction.2
Nørskov et al. stated that the adsorbed oxygen 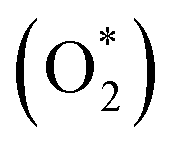 and hydroxyl (OH*) [see reaction scheme depicted in ESI S1†] are found to be very stable intermediates at the equilibrium potential (E° = +1.229 V) for metallic surfaces, and the calculated rate constant for the activated proton/electron transfer to adsorbed oxygen or hydroxyl is directly related to the observed kinetics. According to the volcano plots (Fig. S1†), for the catalytic activity of metals towards ORR, Pt and Pd show the best binding abilities for oxygen and hydroxyl ions.3 Therefore, ever since FCs were developed, such precious metals were the best choice for ORR catalysts, though, Pt has limitations due to its high cost, rareness, low methanol-tolerance and poisoning by carbon monoxide etc., where the latter two are important when used in direct methanol fuel cells.
and hydroxyl (OH*) [see reaction scheme depicted in ESI S1†] are found to be very stable intermediates at the equilibrium potential (E° = +1.229 V) for metallic surfaces, and the calculated rate constant for the activated proton/electron transfer to adsorbed oxygen or hydroxyl is directly related to the observed kinetics. According to the volcano plots (Fig. S1†), for the catalytic activity of metals towards ORR, Pt and Pd show the best binding abilities for oxygen and hydroxyl ions.3 Therefore, ever since FCs were developed, such precious metals were the best choice for ORR catalysts, though, Pt has limitations due to its high cost, rareness, low methanol-tolerance and poisoning by carbon monoxide etc., where the latter two are important when used in direct methanol fuel cells.
Although, there are successful alternatives for anodes that have been developed and commercialized, the commercially cheap cathode is still a conception. Ideal alternative should have a large improvement in ORR catalytic activity and the aim of developing such alternatives is to reduce the Pt-loading or substitution of alternative Pt-free materials in place of Pt. In recent years, considerable improvements have been achieved and nowadays platinum loadings of as low as 0.1 mg cm−2 have shown to have the same performance as that of pure Pt.4 Some researchers have prepared platinum alloys or platinum-free alloys using other transition metals such as palladium(Pd), rhodium(Rd), cobalt(Co), silver(Ag), copper(Cu), gold(Au), nickel(Ni) etc. and these catalysts have performed better than naked platinum as shown in many publications though they also have their inherent problems.5–11 Moreover, the nanoparticles of metals or metal alloys have been prepared with supporting materials to increase the surface area and to minimize metal loading.12–16 During the last few years, metal-free catalysts and catalysts containing only metal cations and supporting cheap materials have been developed as ORR catalysts.17–23 These systems merit the need for more research towards developing cheap alternatives for the oxygen reduction catalyst for the applications in FCs.
Supporting materials are used to disperse the active materials as usable forms for catalytic applications. They should provide some properties such as optimal dispersion of the active component, good accessibility and stability, cheap and readily availability. Moreover, for a good electrocatalyst for ORR, supporting materials should also have a good electronic conductivity, electrochemical and chemical stability and ability to make strong interactions with active materials. In many ORR electrocatalysts, the active component is a small metal cluster, while the support is a hard and porous un-reactive material with considerable electrical conductivity. Porous carbon is the typical example for supporting material and is mostly used in fuel cell applications. Moreover, titania, alumina, montmorillonite (MMT) and silica can also be used as solid supports.2,12,16 However, the latest approach is the use of graphene, carbon nanotubes or organic and hybrid organic/inorganic polymers which can influence catalytic parameters in various ways.8,11,12,15,24–26
We have been pioneered in developing a large number of MMT/electronically conducting polymer/reduced form of the oxidizing cation type of nanocomposites and their applications as very low-cost oxygen reduction electrodes for fuel cells. We have already published two such systems cerium(III)/MMT/polypyrrole (PPY)23 and silver/MMT/PPY.27 In this publication, we discuss preparation, characterization and oxygen reduction catalytic activities of Co(II)/MMT/PPY, Co(II)/MMT/PANI and Co(III)/MT/PEDOT composite systems.
Experimental
Chemicals
Bentonite clay (Sigma), aniline (99.5%, Sigma-Aldrich), pyrrole (99.0%, Sigma-Aldrich), EDOT (97.0% Alfa Aesar), CoK2(SO4)2·6H2O (99.0%, Vickers), acids and bases (VWR Chemicals), H2O2 (30% in water, Sigma-Aldrich), ammonia (30% in water, VWR Chemicals) were used and the chemicals were further purified as and when necessary. For example, bentonite was purified according to the procedure given below and the monomers such as aniline, pyrrole and EDOT were purified by reduced pressure distillation prior to use.Purification of bentonite
Bentonite clay (20.0 g) was added to 0.100 mol dm−3 hydrochloric (HCl) acid solution (800.0 cm3) and stirred for 24 h. The suspension obtained was centrifuged and the supernatant was discarded. Acid-treated clay was added to distilled water (800 cm3) and stirred for 24 h. The suspension obtained was centrifuged and the supernatant discarded. The upper part of the slurry was separated out and re-dispersed in distilled water. This procedure of dispersion, centrifuging and supernatant removal was repeated several times to obtain fine particles of pure MMT. The clay sample thus obtained was dried under regulated air flow under ambient temperature.Preparation of Co(III) ion-exchanged montmorillonite [Co(III)/MMT]
CoK2(SO4)2·6H2O (22.10 g) was dissolved in distilled water (100.0 mL) to prepare 0.50 mol dm−3 Co2+ (aq) solution. Then, 30% NH3 (12.75 mL) and 30% H2O2 (40.0 mL) were simultaneously added drop-wise while stirring the solution in order for Co(II) to get oxidized to Co(III). MMT (1.00 g) was added to 0.500 mol dm−3 Co(III) solution (50.0 mL) and stirred for 48 h. The suspension obtained was centrifuged and the slurry of Co(III)/MMT was collected by discarding the supernatant. The sedimented Co(III)/MMT composite was washed several times with a repeated procedure of dispersing in water, centrifuging and supernatant decanting. The product obtained was dried under ambient conditions and powdered in an agate mortar and pestle. Powdered sample was characterized by XRD, XRF, XPS and FTIR. Then the sample was heated at 150 °C for 2 h in order to remove water present in the interlayer spaces and then again characterized by XRD to obtain interlayer distance when anhydrous cations are present.Preparation of Co(II)/MMT/PANI, Co(II)/MMT/PPY and Co(II)/MMT/PEDOT composites
Co(III)/MMT composite (0.500 g) was dispersed in distilled water (25.0 mL) and the mixture was sonicated for 2 min, until the powdered part is fully dispersed in the solution. Freshly distilled aniline (0.500 g) was dissolved in 1.00 mol dm−3 HCl (aq) solution (25.0 mL) and it was added to the Co(III)/MMT dispersion. The mixture thus obtained was stirred for 48 h and the composite was separated by centrifuging followed by supernatant-discarding and washed with 0.100 M HCl solution and then with acetone. The green coloured precipitate was allowed to dry under ambient conditions and powdered in an agate mortar and pestle to get Co(II)/MMT/PANI composite. In the preparation of black coloured Co(II)/MMT/PPY composite, the same procedure was followed but pyrrole was used instead of aniline. The procedure adapted for the preparation of Co(II)/MMT/PEDOT is slightly different and is outlined below. Co(III)/MMT composite (0.100 g) was dispersed in 0.100 M HCl solution (10.0 mL) and the mixture was sonicated for 2 min until the powdered part is fully dispersed in the solution. Methanol (10.0 mL) was then added. Then 3,4-ethylenedioxythiophene (65 μL) was added and stirred for 48 h. The composite was then separated by centrifuging followed by supernatant-discarding and washed with distilled water and then with acetone. The blue coloured sample was allowed to dry under ambient conditions and powdered in an agate mortar and pestle.Characterization of samples
X-ray diffraction (XRD) analysis was performed to identify the crystalline phases of raw samples and synthesized products and the diffractogrammes were obtained from Siemens D5000 X-ray powder diffractometer with Cu Kα radiation of wavelength, λ = 0.154 nm and at scan rates of 1° or 2° min−1. The XRD patterns were analysed using ICDD PDF 2 database with the aid of X-powder 12 software. The 2θ range 3–15° was chosen for the comparison of d values of interlayer spaces of MMT. Every sample was dried at 150 °C for 2 h and then again XRD patterns were taken to determine thicknesses of interlayer spaces of MMT containing anhydrous cations. The qualitative analyses for elements (those with atomic numbers higher than that of aluminium) of natural and synthetic samples were performed from Fisher X-ray Fluorescence (XRF) instrument for powdered samples. These data are used to compare the atomic ratios of the selected two or more elements that are present in the samples. The morphology and the particle size of products were examined from Hitachi SU6600 Scanning Electron Microscope (SEM) at an acceleration voltage of 10 kV and from Leo 1530 VP field emission gun scanning electron microscope (FE-SEM) at an accelerating voltage of 5 kV. EDX analysis was also done for selected points of SEM images to identify the elements present in the sample and thereby to get an idea about the elemental ratios. FT-IR spectra of powdered samples were examined on a Shimadzu IR prestige 21 instrument with the aid of the KBr pellet method. In the preparation of KBr pellets, the sample was mixed with KBr at the mass ratio of sample![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :KBr of 1:40 and pressed at a pressure of 5 tonnes. The prepared pellets were well dried in a desiccator to remove physically adsorbed water, prior to analyses. Thermogravimetric analysis (TGA) of the samples were performed using Scinco STA N-650 simultaneous thermal analyser at a heating rate of 10 °C min−1, in an air flow, as well as under nitrogen gas purged conditions. X-ray photoelectron spectroscopy (XPS) of synthesized composites were characterized using Axis Ultra DLD X-ray Photoelectron Spectrometer and the data were re-plotted and the de-convolution of bands were done using CasaXPS 2.3 and Origin pro 8.0 Software. Powdered products were pressed at a pressure of 7 tonnes to prepare pellets with the surface area of 0.143 cm2. The thickness was varied as required. The prepared pellets were well dried in a dry desiccator to remove physically adsorbed water prior to analyses. Prepared pellet was sandwiched in two stainless steel rods with same surface area and a voltage of 0.1 V was applied to the two ends of stainless steel rods and the current was measured. Then the conductivity was calculated according to Ohms law. The four-probe conductivity measurements were done according to the method introduced by van der Pauw et al.28 Gold-coated chromium spring contact probes together with low resistivity meter (measuring range 10 μΩ to 2 kΩ) were used to construct the instrument. Voltage was applied into two probes and the current was measured trough the other two probes and the sheet resistances were measured using a low resistivity meter. Same procedure was repeated by applying voltage to latter two probes and measuring current using former two probes.
:KBr of 1:40 and pressed at a pressure of 5 tonnes. The prepared pellets were well dried in a desiccator to remove physically adsorbed water, prior to analyses. Thermogravimetric analysis (TGA) of the samples were performed using Scinco STA N-650 simultaneous thermal analyser at a heating rate of 10 °C min−1, in an air flow, as well as under nitrogen gas purged conditions. X-ray photoelectron spectroscopy (XPS) of synthesized composites were characterized using Axis Ultra DLD X-ray Photoelectron Spectrometer and the data were re-plotted and the de-convolution of bands were done using CasaXPS 2.3 and Origin pro 8.0 Software. Powdered products were pressed at a pressure of 7 tonnes to prepare pellets with the surface area of 0.143 cm2. The thickness was varied as required. The prepared pellets were well dried in a dry desiccator to remove physically adsorbed water prior to analyses. Prepared pellet was sandwiched in two stainless steel rods with same surface area and a voltage of 0.1 V was applied to the two ends of stainless steel rods and the current was measured. Then the conductivity was calculated according to Ohms law. The four-probe conductivity measurements were done according to the method introduced by van der Pauw et al.28 Gold-coated chromium spring contact probes together with low resistivity meter (measuring range 10 μΩ to 2 kΩ) were used to construct the instrument. Voltage was applied into two probes and the current was measured trough the other two probes and the sheet resistances were measured using a low resistivity meter. Same procedure was repeated by applying voltage to latter two probes and measuring current using former two probes.
To carry out the electrochemical studies, working electrode was prepared according to the following procedure. The catalyst (10 μg) was dispersed in ethanol (2.00 mL) and Nafion (10 μL) was added. Then the mixture was sonicated for 10 min in order to obtain a well dispersed mixture which is hereinafter referred to as the catalytic ink. This catalytic ink (10 μL) was deposited on a well cleaned and polished glassy carbon (GC) electrode of 0.384 cm2 active surface area.29,30 This electrode was used as the working electrode and cyclic voltammetry (CV) or linear sweep voltammetry (LSV) was run using Metrohm Potentiostat 101 and 204. Potentials were applied with respect to the saturated calomel electrode (SCE) and a Pt rod was used as the counter electrode. Scan rate used was 50 mV s−1. 0.10 M KOH (aq) or 0.500 mol dm−3 H2SO4 electrolyte solutions were used as required. Cyclic voltammograms were drawn using Nova 1.7 software and were re-plotted using Origin 8.0 pro software. All current densities were normalized to the geometric surface area of the disk electrode. LSV were done at rotating speeds from 600(±5) rpm to 2500(±10) rpm. AC impedance analysis was done using Metrohm Potentiostat 302 and Nyquist plots and Bode plots were drawn using Nova 1.7 software.23,31–33
Results and discussion
XRD analysis
As we have already reported, the cations present in MMT interlayer spaces can be hydrated by one, two, three etc. water molecular layers and the d-spacings vary according to the number of water layers that are present in the hydration sheaths of cations. In ordinary MMT, if Na+ is present in its anhydrous form, then the interlayer spacing value (d value) is approximately 9 Å, and if the Na+ is hydrated by water monolayer, bilayer, three layers and so on, then d values are, respectively, 12 Å, 15 Å, 18 Å and so on where the interlayer thickness expands by ∼3 Å for each additional water layer around the cation. Further, the thickness of the interlayer spaces also depends on the size of the cation in addition to number of water layers the cation is accompanying with it. As such, if the interlayer spaces of MMTs containing different cations are to be compared then the water present within interlayers must be removed. Since the removal of bound water requires heating at a temperature above 100 °C, the comparison can be best done by heating the samples at 150 °C for 2 h. As such, the X-ray diffractogrammes obtained after heating at 150 °C for 2 h, only the diffractions in the range of (100) plane of MMT i.e., in the 2θ range from 4° to 10° for pure MMT, Co(III)/MMT, Co(II)/MMT/PANI, Co(II)/MMT/PPY and Co(II)/MMT/PEDOT are shown in Fig. 1. The d-spacing values obtained corresponding to the (001) plane of these materials are collected in Table 1.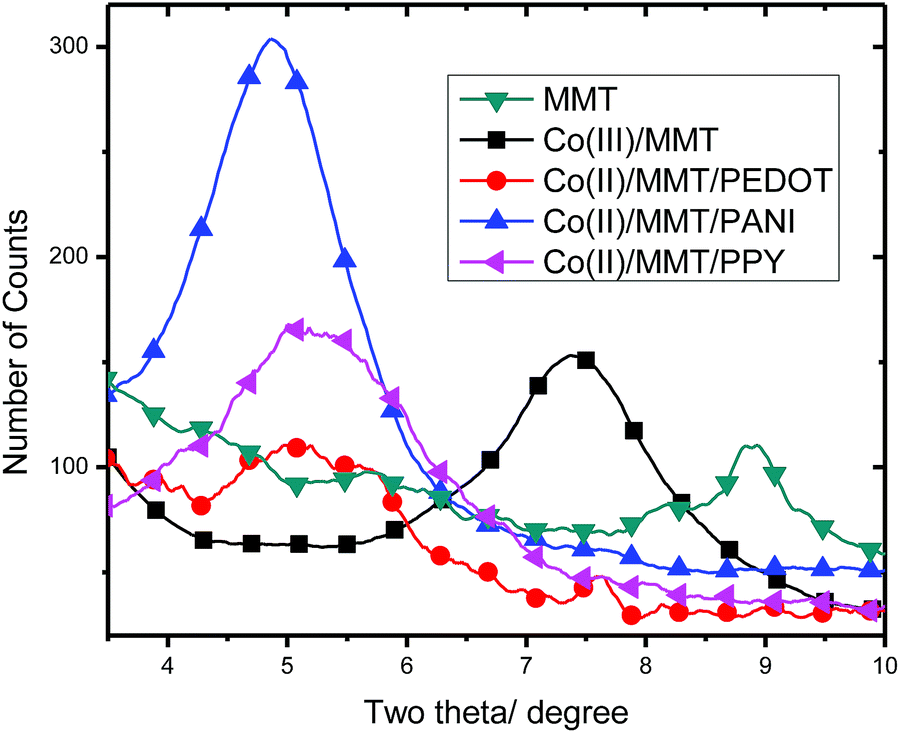 | ||
| Fig. 1 The XRD patterns of montmorillonite [MMT], Co(III)-exchanged montmorillonite [Co(III)/MMT], Co(II)/MMT/PPY, Co(II)/MMT/PANI and Co(II)/MMT/PEDOT, in the 2θ range from 3° to 10° for the comparison of diffractions from (110) planes of the compounds. | ||
| Material | d values/Å | |
|---|---|---|
| At RT | At 150 °C | |
| MMT | 15.2 | 9.62 |
| Co(III)/MMT | 16.2 | 11.8 |
| Co(II)/MMT/PANI | 18.6 | 18.3 |
| Co(II)/MMT/PPY | 17.6 | 17.6 |
| Co(II)/MMT/PEDOT | 18.3 | 18.1 |
Although, the diffractogrammes recorded at room temperature are not shown for the samples, they were indeed recorded, and the corresponding d values are also given in Table 1. It is quite clear from the data depicted in Table 1 that the comparison of d-spaces for MMT containing different species in its interlayers is not possible due to varying amounts of hydration of different cationic species though a good comparison can be made for the samples heat-treated at 150 °C for 2 h. As expected, MMT with anhydrous Na+ gives a d-spacing of 9.62 Å. Tao et al. have studied the swelling of K+, Na+ and Ca2+ montmorillonites and hydration of interlayer cations by a molecular dynamic simulation using CAYFF force field. Their calculations have shown that, even in the anhydrous state of cations, the size of the interlayer spaces depends on the type of cation; for example, in the dry state they have obtained 10.07 Å for Na+-Wyoming and 9.68 Å for Ca2+-Wyoming.34 Similar results have been obtained using Monte-Carlo simulations.35–38 It is interesting to note that Co(III)–MMT has a d-spacing of 11.8 Å and this is because during the preparation of Co(III) from Co(NH3)62+ is converted to [Co(NH3)6]3+ and the actual ions that are present in the interlayer spaces of MMT are [Co(NH3)6]3+ giving rise to a d-spacing of 11.8 Å.
Subsequent discussions of FT-IR and XPS data will show that the ions are bound to alumiosilicate layers of MMT through H-bonding. Introduction of monomers such as aniline or pyrrole or EDOT results in spontaneous polymerisation due to oxidation of monomers by [Co(NH3)6]3+ forming the respective polymers with cobalt in its reduced form of Co(II). This will be confirmed by XPS analysis (vide infra). The interlayer spaces expand so as to accommodate large polymer molecules and Co(II) species. Introduction of conducting polymers makes the interlayer spaces hydrophobic by expelling water even at room temperature (RT). Therefore, the interlayer spaces of Co(II)/MMT/PPY, Co(II)/MMT/PANI and Co(II)/MMT/PEDOT at RT and those of the respective samples treated at 150 °C are the same. The large interlayer spaces indicate the presence of large molecules such as polymers thus giving indirect evidence to the presence of PPY, PANI or PEDOT in respective composites.
As shown in Fig. 1, Co(II)/MMT/PPY has more intense (001) X-ray diffraction than Co(II)/MMT/PEDOT and Co(II)/MMT/PANI has the highest intensity. In order to understand this behaviour, SEM images of the samples were taken. According to the SEM images of Co(II)/MMT/PPY, shown in Fig. 2(a–c), most of the MMT platelets have been fully covered with PPY polymer segments to result in spherically formed PPY polymer matrix around MMT but some MMT layers as plain surfaces can also be seen in these SEM images. The latter are clearly seen in the SEM image shown in Fig. 2(c) where MMT plates and the cross-sections of stacked MMT layers are clearly visible in the encircled region. The SEM images of Co(II)/MMT/PEDOT are shown in Fig. 2(d–f) where phase-separated polymer segments and exfoliated MMT plates together with some polymer intercalated MMT systems are clearly visible. The exfoliation is higher in Co(II)/MMT/PEDOT thus resulting in fewer interlayer spaces of MMT than those in Co(II)/MMT/PPY. This explains why the XRD intensity of (001) plane of MMT in Co(II)/MMT/PEDOT is lesser than that in Co(II)/MMT/PPY.
 | ||
| Fig. 2 SEM images of Co(II)/MMT/PPY (a–c) and Co(II)/MMT/PEDOT (d–f). | ||
XRF analysis
XRF data, shown in Table 2, indicate the presence of Co, Si and Fe in the form of atomic percentages. Although the absolute values are not reliable since XRF cannot detect very light elements and hence percentages are calculated based on measured elements, the intensity ratios of two or more elements in different samples are reliable and can be used for comparison purpose. Co(III)/MMT has 19.78% of Co when compared to other detectable elements. Co:Si atomic ratio of the composites is 0.28. However, the latter amount has been reduced down to 0.21, 0.24 and 0.20, respectively in Co(II)/MMT/PANI, Co(II)/MMT/PPY and Co(II)/MMT/PEDOT composites. This may be due to the removal of some Co(II) ions into the solution during the polymerization process.
| Material | Element% | Ratio | |||
|---|---|---|---|---|---|
| Si | Co | Fe | S | Co/Si | |
| MMT | 70.37 | 0.01 | 29.63 | — | 0.00 |
| Co(III)/MMT | 70.33 | 19.78 | 9.89 | — | 0.28 |
| Co(II)/MMT/PANI | 73.33 | 15.56 | 11.11 | — | 0.21 |
| Co(II)/MMT/PPY | 72.41 | 17.24 | 10.34 | — | 0.24 |
| Co(II)/MMT/PEDOT | 58.26 | 11.91 | 11.57 | 18.26 | 0.20 |
XPS analysis
XPS spectra of Co(III)/MMT composite and Co(II)/MMT/PANI composite do not clearly show the peaks corresponding to the Co 2p3/2, because of low surface concentration of Co. This is due to the fact that cobalt ions are intercalated within the MMT interlayer spaces and are covered by PANI polymer segments as shown by SEM images. Since XPS is a surface analytical technique which analyses the composition within 50 nm thickness from the surface, the less abundance of Co on this region gives rise to weak X-ray photoelectrons.However, the smooth–fit curves for XPS Co 2p2/3 core-level peaks for Co(III)/MMT and Co(II)/PANI/MMT compounds, which are shown in insets of Fig. 3(a) and (b) display the corresponding Co 2p2/3 core-level peaks. Moreover, positions of peaks at 778.4 eV, 779.0 eV, 781.0 eV, and 785.6 eV, can be assigned to be due to Co(0), Co(III), Co(II), and satellite peaks of Co(II) respectively.39,40 According to the peaks of Co(III)/MMT which we have prepared, does actually contain cobalt in its (III) oxidation state. However, paramagnetic Co2+ has a strong shake-up satellite at about 785.6 eV, while the diamagnetic, low-spin Co3+ ion does not show shake-up peaks.41,42 Hence, it can be shown that Co2+ also exists in a small amount. The peaks corresponding to Co2+ are highlighted in the XPS spectrum of Co(II)/MMT/PANI where the increased levels of Co(II) have come from the reduction of Co(III) to Co(II) while aniline is oxidatively polymerized to PANI. As such the polymerization reactions can be expressed as depicted in eqn (2)–(4) and this will be discussed later (vide infra).
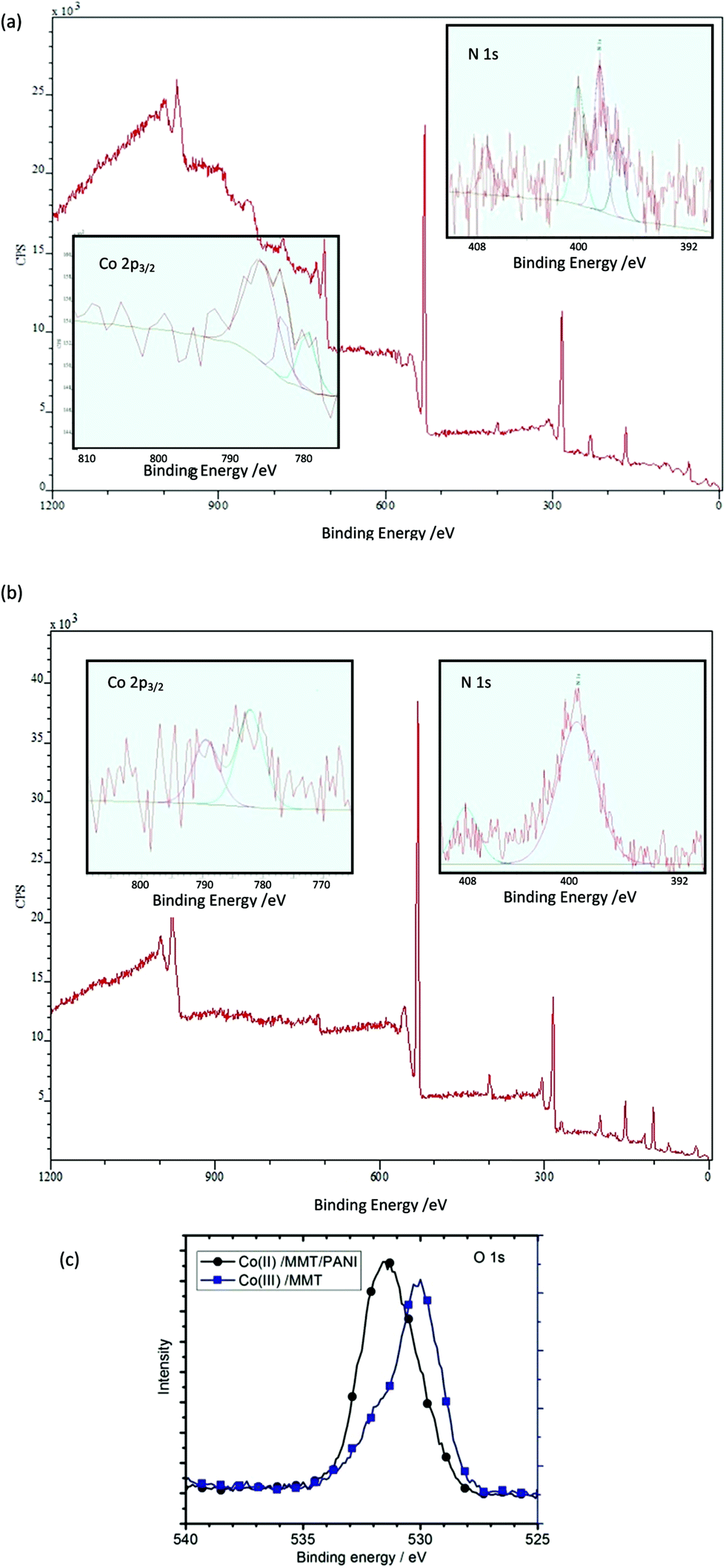 | ||
| Fig. 3 Full range XPS of (a) Co(III)/MMT and (b) Co(II)/MMT/PANI: insets are high resolution XPS of Co 2p3/2 and N 1s (c) XPS of O 1s for both Co(III)/MMT and Co(II)/MMT/PANI. | ||
XPS spectra of N 1s for Co(III)/MMT and Co(III)/MMT/PANI are shown in insets of Fig. 3(a) and (b) respectively. Co(III)/MMT give 3 intense peak at around 401.0 eV, 399 eV and 398 eV which can be ascribed to a quaternary-N of ammonia in the complex of Co(III) has been diminished for the Co(II)/MMT/PANI. The latter shows more intense broad peak at 399.5 eV which corresponds to –NH–C in PANI. Peaks above 405.0 eV are due to the oxidized N such as NO3− ions.41,42
High resolution XPS of O 1s for both Co(III)/MMT and Co(II)/MMT/PANI are shown in Fig. 3(c). The peak corresponding to Si–O–H which appears around 532 eV for O in MMT layers are diminished in Co(III)/MMT due to the formation of coordination bonds with cobalt ions by oxygen on MMT layers making Co–O–Si bonds. However, after the polymerization of aniline, within the interlayers of MMT, the Co–O–Si bonds have been broken to release Co(II) and hence, the usual MMT characteristic spectrum reappears. For XPS peak position of Si–O–H in Na+–MMT is at 532.9 eV but this peak is shifted to 531.5 eV in Co(II)/MMT/PANI.
According to the all characterizations, the following reactions can be suggested for the polymerization process.
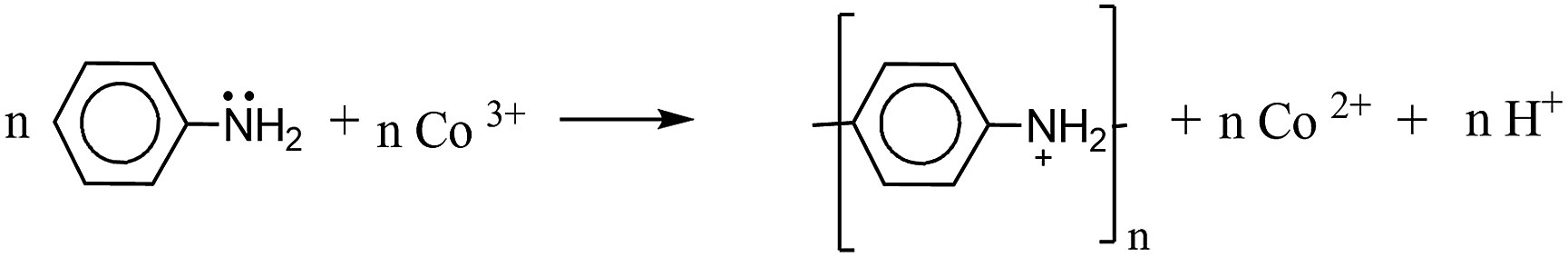 | (2) |
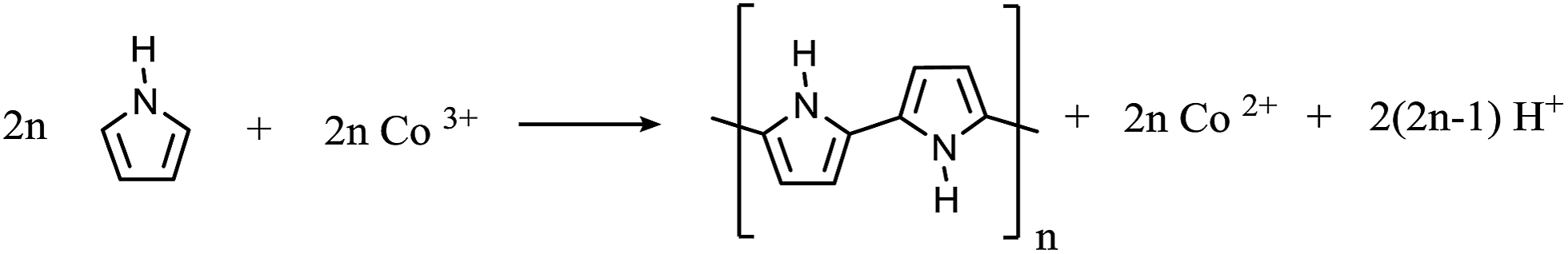 | (3) |
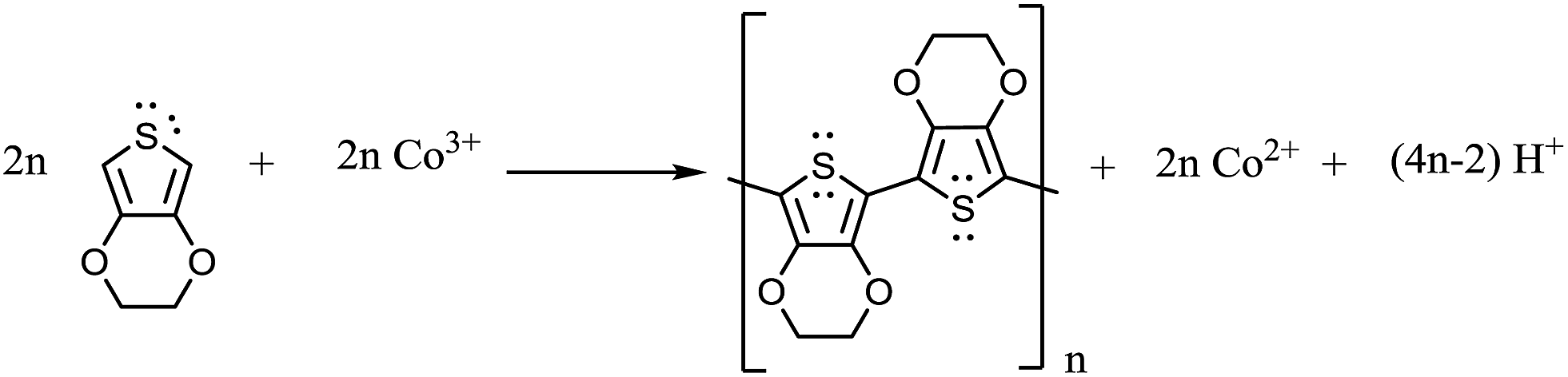 | (4) |
However, Co(III)/MMT composite has [Co(NH3)6]3+ complex ion and [Co(NH3)6]3+ to [Co(NH3)6]2+ reduction has very low standard reduction potential around 0.108 V which is not sufficient to oxidise monomers in to polymer form.43 However, [Co(H2O)6]3+ to [Co(H2O)6]2+ standard reduction potential is very high +1.92 V.43 Hence, the [Co(H2O)6]3+ is a good oxidant which is comparable to persulfate ion. However, it is not stable at ambient conditions. During the reaction, acidic environment converts [Co(NH3)6]3+ to [Co(H2O)6]3+ by removing of ammonia in the complex to ammonium ions and [Co(H2O)6]3+ ions act as the oxidant of the reaction. In order to confirm whether the oxidation of the monomers and subsequent polymerization is taking place through proposed reactions or whether the oxidation is aided by the molecular oxygen, the polymerization reaction was attempted under nitrogen-purged solution. We found that the products obtained in the presence of oxygen were also obtained in the N2-purged solution also. This shows that molecular oxygen is not the oxidant. In order to see whether the polymerization could be done with Co[NH3]63+ the polymerization was attempted in the basic medium at pH 8. We found that the polymerization does not take place in all three cases. Of course, for the polymerization of aniline to conducting emeraldine salt form of polyaniline, an acidic medium is mandatory. However, for the polymerization of pyrrole and EDOT such an acidic medium is not required. Since polymerization did not happen in the basic medium we conclude that Co[NH3]63+ that is present in the interlayer spaces of MMT is incapable of polymerizing aniline, pyrrole or EDOT as expected from thermodynamic considerations through respective electrode potentials. However, when acid is used Co[NH3]63+ is converted to [Co(H2O)6]3+ within the interlayer spaces of MMT which is highly oxidizing and is capable of oxidatively polymerizing these monomers as observed. We also attempted to synthesize [Co(H2O)6]3+ though its instability became a problem. In order to prepare [Co(H2O)6]3+ we oxidized [Co(H2O)6]2+ using ozone at 0 °C but the product obtained was not stable at higher temperatures. On the other hand, the kinetics of monomer polymerization by [Co(H2O)6]3+ at 0 °C was far too slow that we could not obtain polymers at 0 °C.
FTIR analysis
FTIR spectra of pure MMT, Co(III)/MMT, Co(II)/MMT/PANI, Co(II)/MMT/PPY and Co(II)/MMT/PEDOT are shown in Fig. 4. The FTIR spectrum of pure MMT shows the IR absorption bands at 523 cm−1 and 466 cm−1 corresponds to Si–O–Si and Si–O–Al deformation vibrations, respectively. The band at 1031 cm−1 indicates Si–O stretching. Al–Mg–OH deformation, Al–Fe–OH deformation and Al–Al–OH deformation can be assigned to the peaks at 830 cm−1, 877 cm−1 and 915 cm−1 respectively.44 There is a broad band in the range 3200–3500 cm−1 in the FT-IR spectrum of MMT which is due to hydrogen bonded O–H stretching of water molecules inside the MMT layer spaces.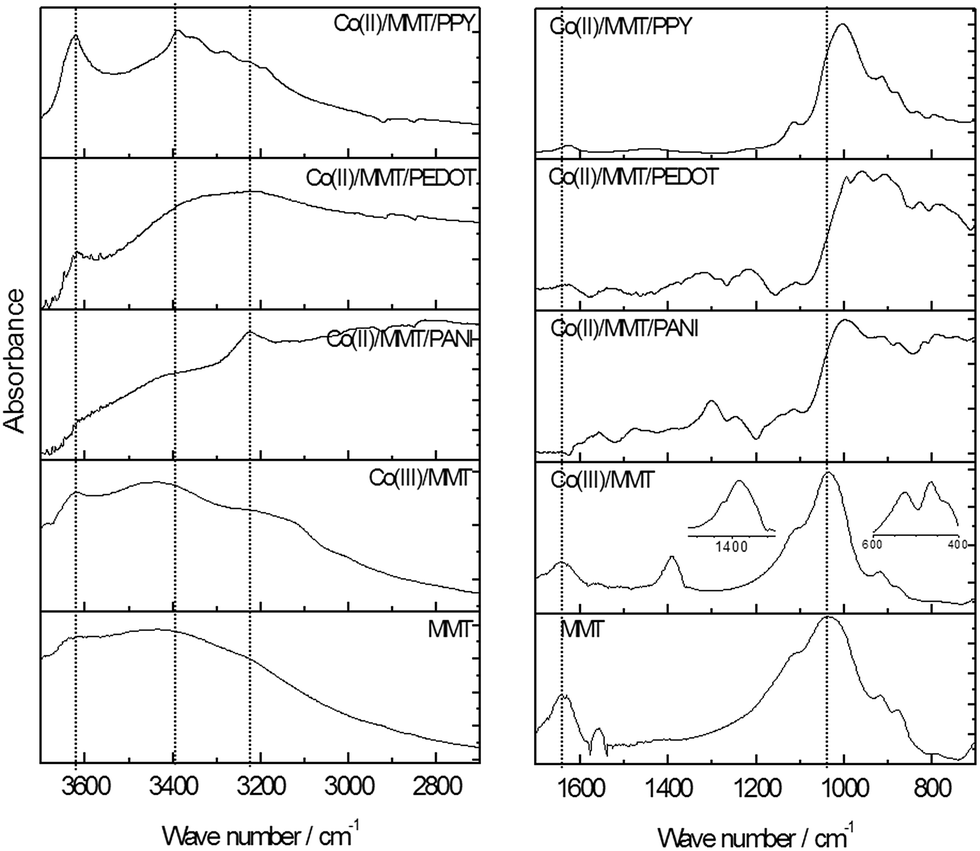 | ||
| Fig. 4 FT-IR spectra of MMT, Co(III)/MMT, Co(II)/MMT/PANI, Co(II)/MMT/PPY and Co(II)/MMT/PEDOT. | ||
The band which is around 3623 cm−1 can be assigned to be due to the interlayer and intralayer OH stretching or non-hydrogen bonded (free) OH stretching of water molecules. The band which is appearing at 1630 cm−1 is due to bending of H–O–H bonds.45 These bands have some considerable noise due to the variation of bonding properties with OH groups of MMT. Peaks at 2343 cm−1 and 2283 cm−1 are due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (12C and 13C carbon isotopes respectively) of CO2 in the atmosphere of the instrument.46 The characteristic bands of MMT is not disappeared or shifted in Co(III) intercalated MMT.
O stretching (12C and 13C carbon isotopes respectively) of CO2 in the atmosphere of the instrument.46 The characteristic bands of MMT is not disappeared or shifted in Co(III) intercalated MMT.
Hence, no structural change to MMT has occurred as a result of ion-exchange. However, a new doublet band is appeared at 1384 cm−1 and at 1400 cm−1 due to the cobalt(III) oxide lattice vibrations47 which shows that Co(III) is coordinated by the O-species of MMT (inset of the figure shows enlarged version of this band). Appearance of Co–N vibration modes at 523 cm−1 and 460 cm−1 (ref. 48) in the Co(III)/MMT (inset of the figure) proves that the cobalt exists as ammonium complex and as oxides also.
Some bands are masked when polymers are formed within the inter-layers of MMT. The band at 1031 cm−1, which is due to the Si–O stretching is shifted when the polymers are intercalated to the inter-layer spaces of MMT. This observation reveals that the strong bonds between polymers and the MMT layers have been formed. Water bending at 1640 cm−1 is disappeared after the incorporation of polymers. All three Co(II)/MMT/conducting-polymer composites show low intensity of this peak and is due to the removal of water inside the layers when the polymerization has taken place as it was also revealed by XRD studies. This answers the question as to why there were no considerable changes in the d spacing of MMT layers shown even after heated at 150 °C for 2 h. Hydrogen bonded O–H stretching at around 3400 cm−1 and free O–H stretching at 3622 cm−1 are narrowed in both Co(II)/MMT/PPY and Co(II)/MMT/PEDOT. In the Co(II)/MMT/PANI composite these peaks are diminished in intensity when compared to the intensity of the new peak appeared at 3226 cm−1 which is due to H-bonded N–H stretching.
DC conductivity measurements and electrochemical impedance analyses
The DC conductivity data of the cobalt/MMT/CP (CP stands for conducting polymer) composites, measured using the 2-probe technique are summarized in Table 3. For Co(II)/MMT/PANI, Co(II)/MMT/PPY and Co(II)/MMT PEDOT composites, the bulk conductivities of the materials were obtained from the AC impedance analyses and the Nyquist plots obtained are shown in Fig. 5(a) together with the suggested equivalent circuit [Fig. 5(b)].| Compound | Resistance/S−1 | Thickness/10−2 × m | Conductivity/S m−1 |
|---|---|---|---|
| Pure MMT | 3.1 × 107 | 0.55 | 1.24 × 10−6 |
| Co(III)/MMT | 8.3 × 105 | 0.67 | 5.64 × 10−5 |
| Co(II)/MMT/PANI | 1.4 × 102 | 0.77 | 3.8 × 10−1 |
| Co(II)/MMT/PPY | 6.1 × 10 | 0.68 | 7.8 × 10−1 |
| Co(II)/MMT/PEDOT | 2.0 × 102 | 0.92 | 3.2 × 10−1 |
| Area of the pellet = 1.43 × 10−4 m2 | |||
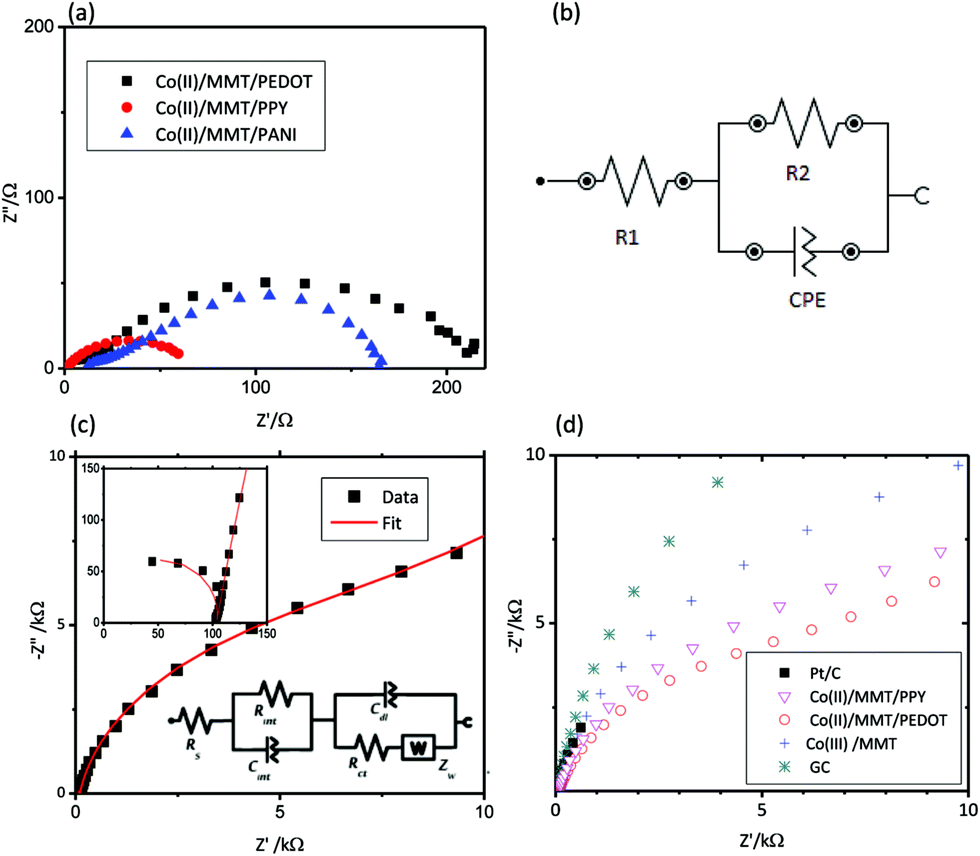 | ||
| Fig. 5 (a) Nyquist plots for composites and (b) suggested equivalent circuit, (c) Nyquist plot and fitted curve for Co/MMT/PEDOT and insets are closer image of high frequency range and corresponding equivalent circuit and suggested equivalent circuit, (d) Nyquist plots of EIS for cobalt composite deposited electrodes together with those of Pt/C in 0.10 M KOH solution at open circuit potentials. | ||
Electrochemical impedance spectra (EIS) of the prepared pellets of composites show a high frequency semicircle as shown in Fig. 5(a) at room temperature, in the frequency range from 0.1 to 1 MHz, under open-circuit conditions. R1 denotes the resistance of the contacts and R2 is the charge transport resistance of the materials. CPE denotes the Constant Phase Element which is not a pure capacitor. Because of this reason, the Nyquist plots are shown distorted semicircles with Z′′ radii lesser than Z′ radii of the circles.
EIS spectrum of Co/MMT/PEDOT composite deposited on GC electrode placed in 0.10 M KOH electrolyte is depicted in the Fig. 5(c). The fitting curve drawn by using the Nova 1.10 fit and simulation analysis is plotted on the same graph in which the inset corresponds to the modified Randles EC. As shown in the figure, the fitting curve matches perfectly well with the data of EIS for Co/MMT/PEDOT. In the EC, Rs denotes the bulk solution resistance, Rint is the interfacial resistance which is attributed to the high-frequency region and Rct is for a charge-transfer resistance at the interface between the electrode surface and the electrolyte, which appears in the low-frequency range. Cint is the inter-facial capacitance Cdl is the double layer capacitance and Zw is Warburg impedance. The serial connection of Rct with the Warburg element and its parallel connection to Rct at low frequency account for the modification. EIS studies were performed for GC, Pt/C, and the three composites (Fig. 5(d)) and the extracted data are summarized in Table 4.
| Compound | Catalyst loading/mg cm−2 | Rint/Ω | Rct/kΩ | Cdl | |
|---|---|---|---|---|---|
| Y0/μS−N | N | ||||
| Pt/C | 0.18 | 84.6 | 12.0 | 1.46 | 0.85 |
| GC | 0.00 | — | 29.0 | 14.0 | 0.98 |
| Co(II)/MMT | 0.18 | 108.2 | 17.92 | 12.9 | 0.83 |
| Co(II)/MMT/PPY | 0.18 | 70.7 | 13.8 | 57.4 | 0.90 |
| Co(II)/MMT/PEDOT | 0.18 | 84.8 | 8.3 | 18.6 | 0.89 |
By studying the electrical elements, which are shown in Table 4, it is clear that Co(II)/MMT/PPY shows the lowest Rint. Since Rint depends on the conductivity of the material the lowest Rint in Co(II)/MMT/PPY accounts for the highest electronic conductivity of the composite among all the materials investigated. When we consider Cct, Co(II)/MMT/PEDOT has considerably lower Cct than that of Pt/C. An ideal catalyst must have low Rct and Rint. Hence, Co(II)/MMT/PEDOT has good catalytic activity which is compatible with that of Pt/C.
Cyclic voltammetry
Electrocatalytic activities have been investigated for the Co(II)/MMT/CP systems by recording their cyclic voltammograms (CVs) in N2 or O2-saturated, 0.10 M aq. KOH solutions using the working electrodes which were prepared by using the composites. Studies were also performed using pristine glassy carbon (GC) electrode, pristine platinum (Pt) electrode and commercially available Pt/C (20% w/w Pt loading) electrocatalysts with the same mass loadings (∼0.18 mg cm−2) for comparison purposes. The CVs obtained are shown in Fig. 6. Fig. 6(a) depicts the CVs for Co(II)/MMT/PANI, GC and Pt electrodes in nitrogen gas saturated environments. According to the figure, all CVs show a current increment at above +0.50 V which is due to the oxidation of hydroxyl ion on the electrode surface in the potential scan in the positive direction. Pt/C shows the highest current gain for the water oxidation. The reduction peaks are observed below −0.30 V for all electrodes. The zoomed images of these reduction peaks are shown in the inset of Fig. 6(a). Cathodic reduction peaks at −0.43 V with a peak current density of 0.72 mA cm−2 is observed for Pt while Co(II)/MMT/PANI electrode shows a small reduction peak at around −0.55 V. However, the current gain is continued until the potential reaches to −1.0 V and at more negative potentials the reduction peak current of Co(II)/MMT/PANI surpasses that of Pt indicating that the former is more efficient electrocatalyst for ORR at more negative potentials.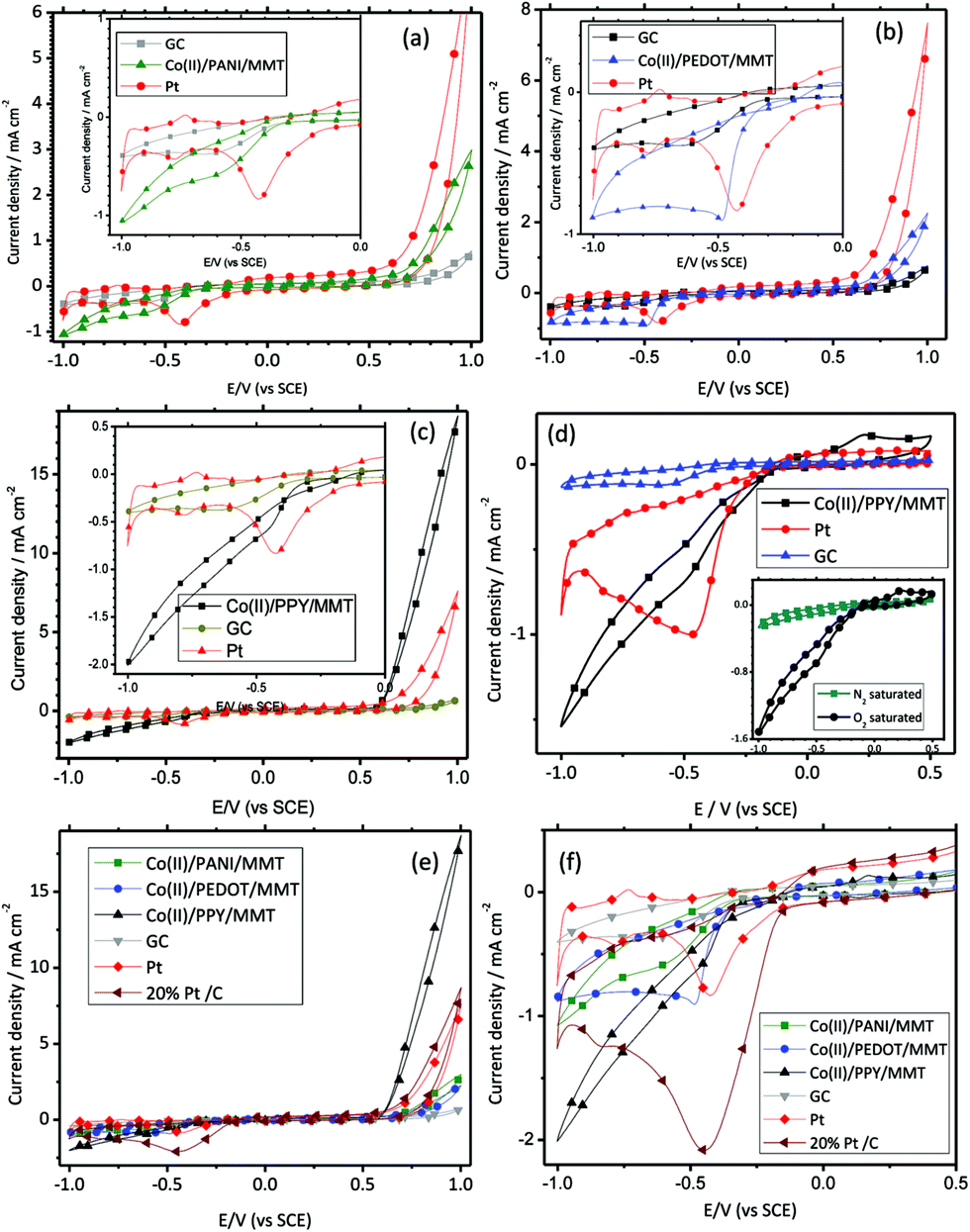 | ||
| Fig. 6 Cyclic voltammogram of (a) Co(II)/MMT/PANI (b) Co(II)/MMT/PEDOT (c) Co(II)/MMT/PPY samples on glassy carbon (GC) electrodes in 0.10 M KOH solution at a scan rate of 50 mV s−1 comparison with the pristine GC and Pt. (d) Lower potential range CVs for Co(II)/MMT/PPY, GC and Pt electrodes in oxygen saturated 0.10 M KOH solution and inset is the comparison of oxygen saturated and nitrogen saturated 0.10 M KOH solutions for Co(II)/MMT/PPY. (e) and (f) CVs for Co-based electrodes and those of GC, Pt/C electrodes in 0.1 M KOH. | ||
Fig. 6(b) displays the comparison of CVs for Co(II)/MMT/PEDOT with those of GC and Pt electrodes in nitrogen saturated environments. According to the figure, all CVs show features as explained above where the current increments above +0.50 V is due to the oxidation of hydroxyl ions. Pt shows the highest current gain for the water oxidation as usual. Hence, more oxygen is produced from Pt electrode. The reduction peaks are observed below −0.30 V for all electrodes. The zoomed image of these reduction peaks are shown in the inset of Fig. 6(b).
For Pt electrode, the reduction peaks at −0.43 V with a peak current density of 0.72 mA cm−2 is observed. The reduction peak at −0.49 V with 0.80 mA cm−2 was observed for the Co(II)/MMT/PEDOT electrode showing that Co(II)/MMT/PEDOT is a better electrocatalyst for ORR than Pt under same mass loading.
Comparisons of CVs for Co/MMT/PPY together with GC and Pt electrodes in nitrogen saturated alkaline environments are depicted in Fig. 6(c) and the inset shows the zoomed image of these reduction peaks. Again the oxidation peaks are observed in all CVs above +0.5 V and are due to the oxidation of hydroxyl ions. Here, the water oxidation on Co(II)/MMT/PPY electrode is better than that on Pt. Hence, more oxygen is produced on the Co(II)/MMT/PPY electrode surface. This may help to increase the oxygen reduction peak current of Co(II)/MMT/PPY simply due to the availability of more oxygen on the electrode surface. Hence, the CVs are taken at lower potential range below +0.5 V since there is no generation of oxygen by the electro-oxidation of water at these potentials. These CVs, carried out in oxygen saturated 0.10 M KOH solutions are shown in Fig. 6(d). In this potential range, from −1.0 V to +0.5 V, reduction peaks are obtained only in the O2 saturated environment while these peaks are absent in the N2 saturated medium, as shown in the inset of Fig. 6(d). For Pt, oxygen reduction peak appears at −0.43 V with a peak current density of 0.72 mA cm−2. The Co/MMT/PPY electrode shows this reduction peak at around −0.48 V but the current gain is continued until the potential reaches −1.0 V. Hence, the catalytic properties towards ORR are higher than that of bulk Pt electrode at larger negative potentials where oxygen is reduced to hydroxyl ions; the reduction half-reaction required for fuel cells. CVs of cobalt-based compounds together with 20% Pt/C, GC and bulk Pt electrodes are shown in the Fig. 6(e) and (f). These results show that the current gains and hence the catalytic activities of the low-cost catalysts that we developed in this work are in between those of bulk platinum and commercial Pt/C catalyst. However the products are very cheap compared to the Pt based catalysts and therefore they are viable competitive alternatives to Pt for developing low-cost fuel cells.
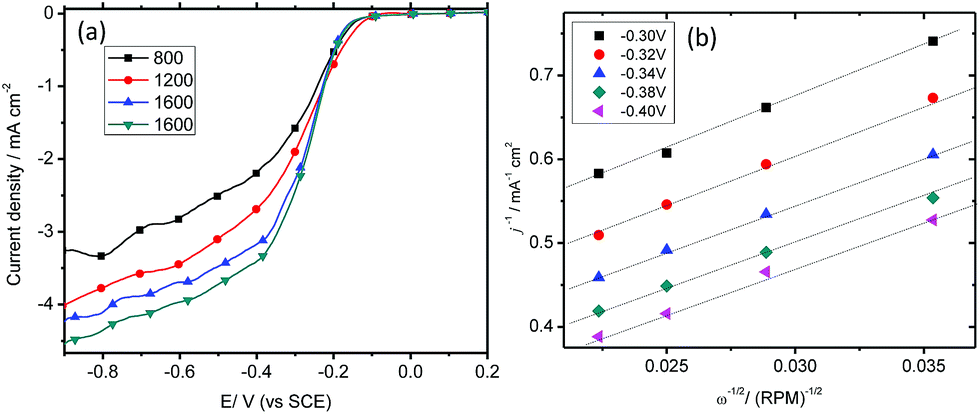
Rotating disk voltammetry
ORR performance is further studied with the linear sweep voltammetry (LSV) measurements on a rotating disk electrode (RDE) of Co-based electrodes, along with the naked GC and Pt/C electrodes, in O2 saturated 0.10 M KOH solutions at a scan rate of 5 mV s−1 and a rotation rate of 1600 rpm. As shown in Fig. 7, the onset potentials for oxygen reduction at Pt/C, Co(II)/MMT/PEDOT, Co(II)/MMT/PPY and GC are −0.11 V, −0.18 V, −0.20 V, and −0.24 V, respectively. The limiting diffusion current density at −0.20 V for the Pt/C, Co(II)/MMT/PEDOT, Co(II)/MMT/PPY and GC electrodes are 1.4, 0.7, 0.4 and 0.07 mA cm−2 respectively. But at −0.40 V, these current values are changed to 4.6, 3.5, 2.1 and 0.5 mA cm−2 respectively. These values are further changed to 5.5, 5.3, 4.8 and 1.5 mA cm−2 respectively at −1.0 V.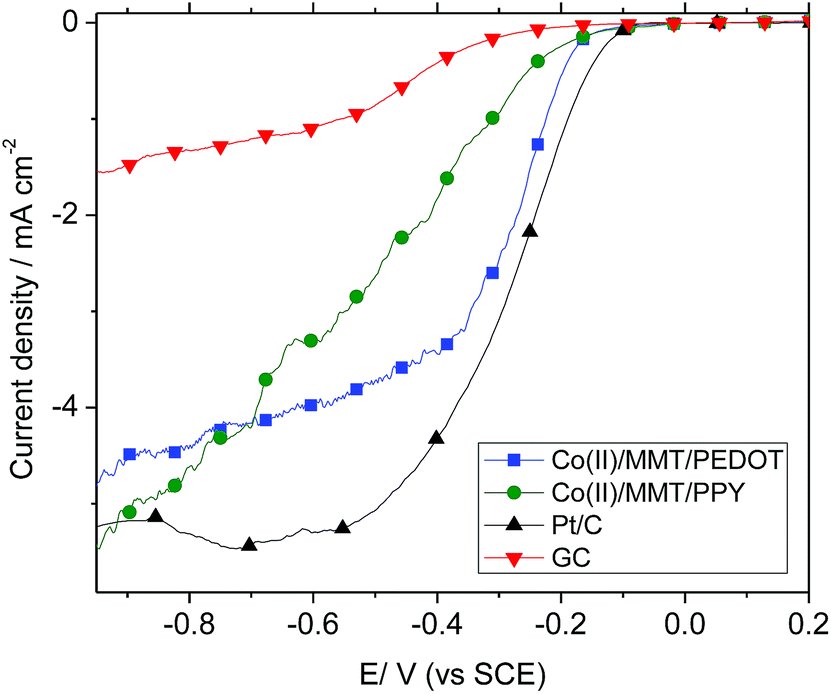 | ||
| Fig. 7 Polarization curves of Co based MMT/polymer composites together with those of Pt and GC electrodes at a rotation rate of 1600 rpm and a scan rate of 5 mV s−1. | ||
Hence, at the large overpotential values, both Co(II)/MMT/PEDOT and Co(II)/MMT/PPY act as competitive catalyst to Pt electrode. These data are consistent with the CV data and confirm the significant contributions to the ORR electrocatalytic activity of Co based compounds. The catalytic activity and kinetics of Co(II)/MMT/PPY toward ORR were analyzed using RDE.
Polarization curves for different rotation speeds are shown in Fig. 8(a). The current density values at −1.0 V are plotted against square root of rotation speed as shown in Fig. 8(b).
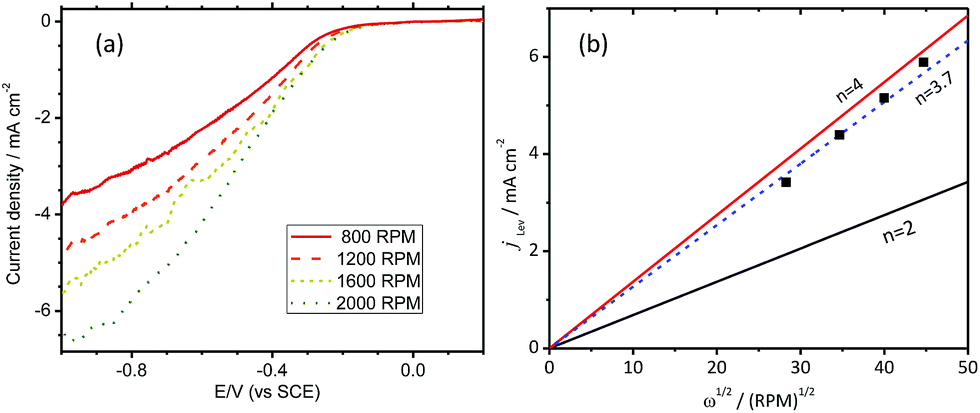 | ||
| Fig. 8 (a) ORR polarization curves of Co(II)/MMT/PPY obtained at different rotating rates. (b) The plots of current density at −1.0 V vs. square root of angular velocity for the systems. | ||
Hence, the obtained data set fit with the graph drawn for an electron transfer number of 3.7 according to the Levich equation (eqn (5)).
| jlev = 0.620nFD2/3ν−1/6ω1/2Cbulk | (5) |
Koutecky–Levich plots, drawn for different voltage values, which were obtained from their polarization curves are shown in the Fig. 9(a). By using the Koutecky–Levich plots, the number of electrons transferred (n) through the Co(II)/MMT/PPY electrocatalytic reaction can then determined using the slop of the plots. Then n vs. potential is plotted and are shown in Fig. 9(b), and the inset is the Koutecky–Levich plots in the entire range. This result reveal that “n” is equal to 2 at the potential below −0.5 V. At this potential range, intercept at this region can be used to determine the kinetic current.
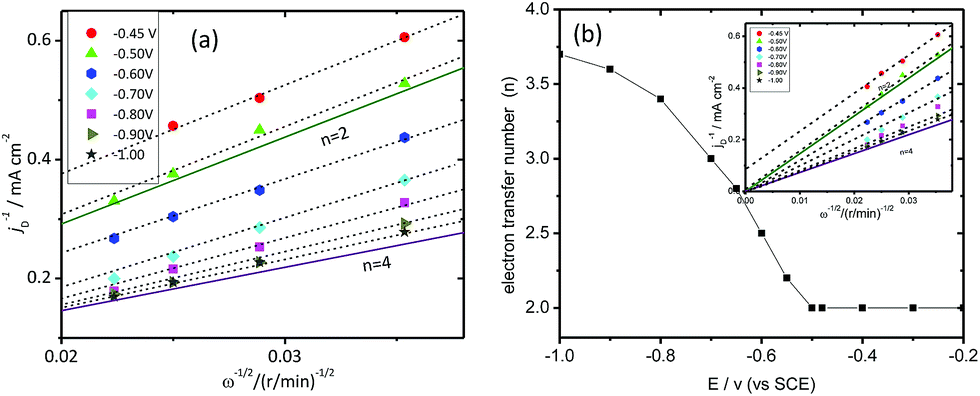 | ||
| Fig. 9 (a) Koutecky–Levich plots of Co(II)/MMT/PPY at different potentials, (b) the plot of electron transfer number calculated from Koutecky–Levich plots vs. potential. | ||
From −0.5 V up to −1.0 V, n is increased and the intercept of the graphs are almost zero and behave according to the Levich equation (eqn (5)). Hence, the material shows good catalytic properties at the higher over potential values. The catalytic activity and kinetics of Co(II)/MMT/PEDOT towards ORR was analyzed using RDE. Polarization curves for different rotation speeds are shown in the Fig. 10(a). Koutecky–Levich plots, drawn for different voltage values which were obtained from their polarization curves, are shown in Fig. 10(b). By using the Koutecky–Levich plots, the number of electrons transferred (n) through the Co(II)/MMT/PEDOT electrocatalytic reaction was then determined using the slop of the plots and the kinetic currents were obtained using the intercept of the graphs. The plots are parallel to each other hence all has same n value coincident with 3.2. Hence the material shows good catalytic properties even at lower over potential values.
 | ||
| Fig. 10 (a) Polarization curves of Co(II)/MMT/PEDOT obtained at different rotating rates in oxygenated 0.10 M KOH (aq) solutions. (b) Koutecky–Levich plots of Co(II)/MMT/PEDOT at different potentials. | ||
Conclusions
The Co(II)/MMT/PANI, Co(II)/MMT/PPY and Co(II)/MMT/PEDOT composites were prepared and characterized for their morphological and chemical behaviours as well as electrical conducting properties and electrocatalytic properties towards oxygen reduction half-reaction. XRD, FTIR, XRF, and SEM data prove that the polymers are intercalated in the MMT layers in Co(II)/MMT/PANI and Co(II)/MMT/PPY composites and Co(II)/MMT/PEDOT has exfoliated structure. XPS data confirms the presence of the Co(III) ions in Co(III)/MMT which then participate in the polymerization and the most Co(III) ions are then reduced to Co(II). Electrochemical analysis shows that both Co(II)/MMT/PPY and Co(II)/MMT/PEDOT have good catalytic activity towards ORR and at higher overpotential values but very interestingly Co(II)/MMT/PPY has competitive performance with commercially available Pt/C even at low overpotentials.Acknowledgements
Authors gratefully acknowledge the financial assistance by Higher Education for Twenty First Century (HETC) Grant (Grant No. HETC/PGIS/QIG/W3-TOR10). We also thank Professor Masaru Shimomura and his research students at the Shizuoka University, Japan for recording XPS spectra.Notes and references
- X. Wei, Y. Geping and Z. Jiujun, Rotating electrode methods and oxygen reduction electrocatalysts, Elsevier, Oxford, 2014, vol. 3, pp. 81–86 Search PubMed.
- G. Rothenberg, Catalysis: Concepts and Green applications. Wiley-VCH, Weinheim, 2008 Search PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem., 2004, 108, 17886–17892 CrossRef.
- G. J. K. Acres, J. C. Frost, G. A. Hards, R. J. Potter, T. R. Ralph, D. Thompsett, G. T. Burstein and G. J. Hutchings, Catal. Today, 1997, 38, 393–400 CrossRef CAS.
- C. Wang, N. M. Markovic and V. R. Stamenkovic, ACS Catal., 2012, 2, 891–898 CrossRef CAS.
- S. P. Lin, K. W. Wang, C. W. Liu, H. S. Chen and J. H. Wang, J. Phys. Chem. C, 2015, 119, 15224–21523 CAS.
- R. Wang, C. Xu, X. Bi and Y. Ding, Energy Environ. Sci., 2012, 5, 5281–5286 CAS.
- F. Fouda-Onana, S. Bah and O. Savadogo, J. Electroanal. Chem., 2009, 636, 1–9 CrossRef CAS.
- B. Li and J. Prakash, Electrochem. Commun., 2009, 11, 1162–1165 CrossRef CAS.
- A. Holewinski, J. C. Idrobo and S. Linic, Nat. Chem., 2014, 6, 828–834 CrossRef CAS PubMed.
- R. E. Cid, J. L. G. Fuente, S. Rojas, J. L. G. Fierro and P. Ocon, ChemCatChem, 2013, 5, 3680–3689 CrossRef.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 8618 Search PubMed.
- L. Perini, C. Durante, M. Favaro, V. Perazzolo, S. Agnoli, O. Schneider, G. Granozzi and A. Gennaro, ACS Appl. Mater. Interfaces, 2015, 7, 1170–1179 CAS.
- B. Li, Z. Yana, Q. Xiao, J. Dai, D. Yang, C. Zhang, M. Cai and J. Ma, J. Power Sources, 2014, 270, 201–207 CrossRef CAS.
- C. Jeyabharathi, P. Venkateshkumar, J. Mathiyarasu and K. L. N. Phani, J. Electrochem. Soc., 2010, 157, B1740–B1745 CrossRef CAS.
- M. Xia, W. Ding, K. Xiong, L. Li, X. Qi, S. Chen, B. Hu and Z. Wei, J. Phys. Chem. C, 2013, 117, 10581–10588 CAS.
- R. Silva, D. Voiry, M. Chhowalla and T. Asefa, J. Am. Chem. Soc., 2013, 135, 7823–7826 CrossRef CAS PubMed.
- Q. He, T. Mugadza, G. Hwang and T. Nyokong, Int. J. Electrochem. Sci., 2012, 7, 7045–7064 CAS.
- J. Ma, X. Wang and X. Jiao, Int. J. Electrochem. Sci., 2012, 7, 1556–1563 CAS.
- Z. Yao, H. Nie, Z. Yang, X. Zhou, Z. Liu and S. Huang, Chem. Commun., 2012, 48, 1027–1029 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 4394–4403 CrossRef CAS PubMed.
- J. Zhang, D. He, H. Su, X. Chen, M. Pan and S. Mu, J. Mater. Chem. A, 2014, 2, 1242–1246 CAS.
- R. M. G. Rajapaksea, K. Murakami, H. M. N. Bandara and R. M. M. Y. Rajapakse, Electrochim. Acta, 2010, 55, 2490–2497 CrossRef.
- V. J. Gillet, P. Willett and J. Bradshaw, J. Chem. Inf. Comput. Sci., 1997, 37, 731 CrossRef CAS.
- J. Beckers, L. M. van der Zande and G. Rothenberg, ChemPhysChem, 2006, 7, 747 CrossRef CAS PubMed.
- B. Winther-Jensen, O. Winther-Jensen, M. Forsyth and D. R. Macfarlane, Science, 2008, 321, 671–674 CrossRef CAS PubMed.
- R. M. G. Rajapakse, K. G. C. Senarathna, A. Kondo, P. S. Jayawardena and M. Shimomura, Advances in Automobile Engineering, 2015, 4, 121 Search PubMed.
- L. J. Van der Pauw, Method of measuring the resistivity and hole coefficient on lamellae of arbitrary shape, Philips Tech. Rev., 1958, 20, 220–224 Search PubMed.
- Z. Yao, H. Nie, Z. Yang, X. Zhou, Z. Liu and S. Huang, Catalyst-free synthesis of iodine-doped graphene via a facile thermal anneal process and its use for electrocatalytic oxygen reduction in an alkaline medium, Chem. Commun., 2011, 45, 1027–1029 Search PubMed , ESI.†.
- R. E. Cid, J. L. G. de la Fuente, S. Rojas, J. L. G. Fierro and P. O. Esteban, ChemCatChem, 2013, 5, 3680–3689, DOI:10.1002/cctc.201300448.
- M. Xia, W. Ding, K. Xiong, L. Li, X. Qi, S. Chen, B. Hu and Z. Wei, J. Phys. Chem. C, 2013, 117, 10581–10588 CAS.
- Z. Jiang, Z. Jiang, X. Tian and W. Chen, J. Mater. Chem. A, 2014, 2, 441 CAS.
- H. Pang, C. Wei, X. Li, G. Li, Y. Ma, S. Li, J. Chen and J. Zhang, Microwave-assisted synthesis of NiS2 nanostructures for supercapacitors and cocatalytic enhancing photocatalytic H2 production, Sci. Rep., 2014, 4, 3577, DOI:10.1038/srep03577.
- L. Tao, T. Ziao-Feng, Z. Yu and G. Tao, Chin. Phys. B, 2010, 10, 109101–109107 CrossRef.
- V. M. C. Paez and K. Workum, J. Chem. Phys., 2001, 114, 1405 CrossRef.
- L. Pablo De, M. L. Chávez, A. K. Sum and J. J. De Pablo, J. Chem. Phys., 2004, 120, 939 CrossRef PubMed.
- E. S. Boek, P. V. Coveney and N. T. Skipper, J. Am. Chem. Soc., 1995, 117, 12608 CrossRef CAS.
- M. Chávez-Páez, L. Pablo De and J. J. De Pablo, J. Chem. Phys., 2004, 114, 24 Search PubMed.
- M. Li, X. Bo, Y. Zhang, C. Han, A. Nsabiman and L. Guo, J. Mater. Chem. A, 2014, 2, 11680 Search PubMed.
- K. Lee, L. Zhang, H. Lui, R. Hui, Z. Shi and J. Zhang, Electrochim. Acta, 2009, 54, 4704–4711 CrossRef CAS.
- M. Oku and Y. Sato, Appl. Surf. Sci., 1992, 55, 37–41 CrossRef CAS.
- Y. Chen, S. Zhao and Z. Liu, Phys. Chem. Chem. Phys., 2015, 17, 14012–14020 RSC.
- D. D. Lide, CRC Handb. Chem. Physics, 87th edn, 2005, pp. 1–10 Search PubMed.
- P. Bala, B. K. Samantaray and S. K. Srivastava, Bull. Mater. Sci., 2000, 23, 61–67 CrossRef CAS.
- M. Chaplin, Water Absorption Spectrum, Water Structure and Science. 2008 Search PubMed.
- H. Yamada and W. B. Person, J. Chem. Phys., 1964, 41, 2478 CrossRef CAS.
- F. Khatun, M. A. Gafur, M. S. Ali, M. S. Islam and M. A. R. Sarker, J. Sci. Res., 2014, 6, 217–231 CAS.
- J. Lichty, S. M. Allen, A. I. Grillo, S. J. Archibald and T. J. Hubin, Inorg. Chim. Acta, 2004, 357, 615–618 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra23100d |
| This journal is © The Royal Society of Chemistry 2016 |