
Designing Ga(iii)-containing hydroxyapatite with antibacterial activity†
Abstract
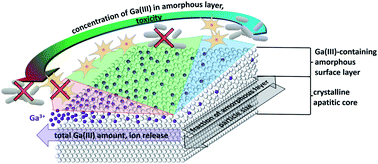
Modified hydroxyapatite with antibacterial properties and high biocompatibility with mammalian cells is very desirable in many biomedical applications. Here we study the incorporation of Ga3+ ions into hydroxyapatite to achieve such an effective biological response. Antibacterial action against P. aeruginosa and cytocompatibility with L929 mouse fibroblasts was examined in vitro for different hydroxyapatites with incorporated Ga3+ ions (HAp(Ga)). Different Ga(III) contents and HAp(Ga) morphologies were obtained by adding Ga3+ ions at different stages of the hydroxyapatite synthesis and varying the Ga3+ concentration. Ga3+ ions were incorporated into the hydroxyapatite precursor phases at the early stages of the hydroxyapatite synthesis and into the amorphous surface layer of the apatite crystals at the late stages or after the synthesis, which yielded a mixed Ca/Ga amorphous phosphate surface layer. Their incorporation into the precursor nuclei before crystal formation and growth reduced the size of the final HAp(Ga) to small apatite nanocrystals with a large fraction of the mixed Ca/Ga amorphous phosphate surface layer. The release of Ga3+ ions from the HAp(Ga) depended on the overall amount of Ga in the material, while the biological response was mediated by the composition of the amorphous surface layer. The mainly Ca-phosphate amorphous surface layer resulted in a low antibacterial efficiency, while the mainly Ga-phosphate amorphous surface layer resulted in a high antibacterial efficiency, but also cytotoxicity. Ga(III)-containing hydroxyapatite can be designed for an effective biological response by varying the HAp(Ga) size and the overall Ga content to achieve the desired composition of the amorphous surface layer. The optimum HAp(Ga) in this study contained 4 wt% of Ga(III) and was obtained by adding Ga3+ ions during transformation of the precursor octacalcium phosphate/hydroxyapatite crystals to the final hydroxyapatite crystals. It provides excellent control over the release of the Ga3+ ions, and joins together the high efficacy of the antimicrobial action and non-toxicity over a wide concentration range (from 0.3 g L−1 to at least 1 g L−1).
 Please wait while we load your content...
Please wait while we load your content...

