
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface modification and porosimetry of vertically aligned hexagonal mesoporous silica films†‡
Calum
Robertson
a,
Andrew W.
Lodge
a,
Peter
Basa
b,
Marina
Carravetta
a,
Andrew L.
Hector
 *a,
Reza J.
Kashtiban
c,
Jeremy
Sloan
c,
David C.
Smith
d,
Joseph
Spencer
d and
Alain
Walcarius
e
*a,
Reza J.
Kashtiban
c,
Jeremy
Sloan
c,
David C.
Smith
d,
Joseph
Spencer
d and
Alain
Walcarius
e
aChemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK. E-mail: A.L.Hector@soton.ac.uk
bSemilab Semiconductor Physics Laboratory Co. Ltd., Prielle Kornélia str. 2, H-1117 Budapest, Hungary
cDepartment of Physics, University of Warwick, Coventry CV4 7AL, UK
dPhysics and Astronomy, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
eLaboratoire de Chimie Physique et Microbiologie pour l'Environnement, UMR 7564 CNRS – Université de Lorraine, 405, rue de Vandoeuvre, 54600 Villers-les-Nancy, France
First published on 11th November 2016
Abstract
Mesoporous silica films with vertically aligned hexagonal pores have been produced via the electrochemically assisted surfactant assembly (EASA) method using cetyltrimethylammonium bromide (CTAB) surfactant. Mesoporous silica powder has also been synthesised using the same surfactant. The pore walls of the silica powder and films have been grafted with organosilane reagents. The size of the pore, degree of grafting and effect on the properties of the pore have been investigated using porosimetry, contact angle, NMR and CHN analysis. The degree of grafting was found to be dependent upon the size of the grafting agent, with the smallest steric bulk grafting most effectively. It was found that the grafting of the pores with Me3SiCl greatly increased the hydrophobicity of the pore and reduced water penetration. Grafting with larger groups caused the film surface to be hydrophobic but had little effect on the penetration of water into the pores.
Introduction
Mesoporous silica powders can readily be produced with various pore sizes and with 1D or 3D pore structures, and hence provide a chemically stable platform for applications in catalysis, adsorption and sensors.1–4 An important aspect of their utility is the ability to change the surface chemistry of the pores by addition of reagents containing direct Si–R bonds (R = substituent) to the silica synthesis procedure.5–8 An early example of modification of the pore walls after synthesis of the silica (“grafting”) used Me3SiCl to replace the hydroxyl groups on the pore walls with trimethylsilyl groups, causing a decrease in the diameter of the pore.9 This also replaces the relatively hydrophilic silica surface with a hydrophobic one terminated by C–H bonds. Using substituents that contain functional groups it has also been possible to produce selective catalysts,10 sensors for humidity, pH and metal ions,11 proton conductors,12 electrochemical sensors13 and biosensors,14 and absorbents to remove contaminants from water15,16 or air.17Mesoporous silica films are commonly synthesised via the evaporation induced self-assembly (EISA) method,18–21 using solutions of tetraethyl orthosilicate (TEOS) and a surfactant in a water/ethanol sol. In the EISA method, the sol is dip-coated onto the substrate and as the ethanol evaporates the surfactant concentration increases, causing the formation of micelles, around which the silicates condense. The pore diameter of the resultant silica is determined by the size of the micelles, and their orientations by interactions between micelle surfaces and neighbouring micelles or the substrate. Vertical alignment is very difficult to achieve, but with cationic surfactants such as cetyltrimethylammonium bromide (CTAB) hexagonally packed arrays of vertically aligned mesopores can be obtained by the electrochemically assisted surfactant assembly (EASA) method22–27 (alternatives are Stöber solution growth or oil-induced co-assembly methods28,29). EASA uses an electronically conductive substrate immersed in the sol. The application of a negative potential causes the self-assembly of the cationic surfactant and the passing of the current results in the formation of hydroxide ions close to the substrate surface.26,30,31 This pH change catalyses the condensation of the silicate and porous silica structures form around the micelles.32 For both methods, once the porous structure has been formed the surfactant is removed by pyrolysis or Soxhlet extraction leaving empty pores.27 EASA films have been suggested for applications as electrochemical sensors, biosensors and adsorbents.13,14,16
Our interest in surface modification of EASA films is related to the possibility of using them as templates for supercritical fluid electrodeposition to grow arrays of very small diameter nanowires.27,33–35 Supercritical fluids can effectively be used to penetrate small pores due to their low viscosities and zero surface tension. Previously we showed that EASA films on titanium nitride, a substrate chosen for its compatibility with a wide range of electrodeposition conditions34,36 and with contacting of many functional nanowire materials, results in pore diameters of ∼1.6 nm.27 Grafting of these pores may provide changes in pore size, access of dissolved species to the electrodeposition growth surface or modifications to the properties of nanowires grown in the templates. Previous work on the modification of EASA films has involved co-condensation of TEOS with an organosilane26,30,31 or coupling this approach to “Click” chemistry to modify the silica surface.37,38 Post synthesis functionalisation has also been demonstrated with hydroquinone derivatives.39 In this paper, the effects of several grafting agents on the properties of EASA silica films is examined, with studies of the same chemistries on mesoporous silica powders used to provide further insight.
Experimental
A one-pot synthesis of mesoporous silica powder was derived from the work of Nooney et al.4 A solution of 40.7 cm3 aqueous ammonia (37% w/w, Fisher) in 442 cm3 H2O was heated to 50 °C whilst stirring in a round-bottomed flask. CTAB (1.10 g, Sigma-Aldrich) was dissolved in the solution, TEOS (5.6 cm3, 98%, Sigma-Aldrich) was added, and the solution was stirred whilst being allowed to cool to room temperature over two hours. The precipitate was filtered under suction and the remaining powder was dried at 60 °C for 16 hours. The surfactant was removed from the silica by washing the powder in a Soxhlet extraction thimble with 0.1 M HCl/EtOH for 6 hours.MOCVD titanium nitride coated silicon wafers were purchased from Si-Mat. The wafers were supplied with a PVD silica capping layer and a 0.8 cm diameter circular region was etched into the silica using aqueous HF to provide a deposition surface with the remainder of the electrode insulated by the silica film. Mesoporous silica films were produced on TiN films using the electrochemically assisted surfactant assembly (EASA) method. A 10 mM solution of NaNO3 in H2O (20 cm3) was added to 20 cm3 of ethanol. 0.2 M HCl/H2O was added dropwise to the solution until the pH was approximately 3. TEOS (0.905 cm3) was added to the solution and allowed to stir for 90 minutes. CTAB (0.47 g, 1.29 × 10−3 mol) was added to the solution and allowed to stir for 30 minutes until fully dissolved. A cone shaped PTFE electrochemical cell was equipped with a steel counter electrode, a 3 mm diameter silver rod pseudo-reference electrode, and the TiN coated Si wafer working electrode. A potential of −1.3 V vs. Ag/Ag+ was applied for 20 s. The films were immediately removed from the solution and washed with water before being dried in air overnight at 130 °C. The CTAB surfactant was removed from the mesopores by Soxhlet extraction over 5 h using 0.2 M HCl/EtOH.
For grafting, the mesoporous silica powder (0.5 g), or an EASA silica film on a silicon/TiN wafer, was first dried at 110 °C under vacuum for 6 h then handled under nitrogen to avoid introducing excess moisture that could react with the grafting agents to block pores. The grafting agents that were used were trimethylchlorosilane (Me3SiCl), phenyldimethylchlorosilane (PhMe2SiCl), tert-butyldimethylchlorosilane (tBuMe2SiCl) and hexamethyldisilazane (HN(SiMe3)2), and all were obtained from Sigma-Aldrich and used as supplied. The powder or film was placed in 10 cm3 of dry THF and 3.9 mmol of the grafting agent was added, followed by refluxing for 24 h. The solution was removed and the sample was washed with THF (5 × 20 cm3) and diethyl ether (5 × 20 cm3) followed by Soxhlet extraction for 6 hours with diethyl ether followed by drying under vacuum at 110 °C.
Small angle X-ray scattering (SAXS) was performed on a Rigaku Smartlab Thin Film instrument using a 9 kW Cu-Kα source and a DTex250 1D detector. Powder samples were mounted inside 0.7 mm diameter borosilicate glass capillaries and measured from 1° to 10° 2θ. For thin films, grazing incidence (GI)SAXS scans were collected from 1 to 10° 2θ using a 0.25° incidence angle and either in-plane or out-of-plane geometry. Transmission electron microscopy (TEM) was conducted in a JEOL 2100 LaB6 instrument operating at 200 kV.
29Si MAS-NMR measurements were performed on a wide bore 14.1 T Bruker Avance II spectrometer on a 4 mm probe with a spinning frequency of 8 kHz. The spectra were acquired using cross polarisation (CP) and 1024 scans per sample, and referenced using silicone rubber at −22.3 ppm as indirect reference. Additional 29Si data were acquired using direct excitation with a pulse delay of 600 s, and in the Me3Si-grafted sample pulse delays of 900 s were also used to check that sites were fully relaxed and hence that the relative intensities of features were fairly quantitative. 13C MAS NMR data were acquired using 1024 CP scans. The axis was calibrated using adamantane at −38.5 ppm as indirect reference.
Nitrogen adsorption–desorption measurements were performed on powder samples using a Micromeritic 3Flex Surface Characterisation Analyser. The powder samples were dehydrated prior to measurement by heating to 110 °C under vacuum overnight to remove any water. The samples were analysed at 77 K. Surface area was determined by the Brunauer–Emmett–Teller (BET) method.40 The pore size distribution was modelled using a non-local density-functional theory (NLDFT) approach41–44 implemented within the Micromeritics SAIEUS software package (Solution of Adsorption Integral Equation Using Splines). Porosimetry on thin film samples used a PS-2000 ellipsometric porosimeter, with samples exposed to water, toluene, isopropanol or methanol vapour with ellipsometry incidence angles of 60°. The BET method was again used to model surface area, with a modified Kelvin equation or the Dubinin–Rasushkevich model used to calculate the pore size distribution.45,46 The substrate contribution was modelled on a plain wafer of MOCVD TiN-coated silicon.
Water contact angle measurements were made using digital photographs of a 1 μL water droplet on the surface. Combustion analysis (for C, H and N) was outsourced to Medac Ltd. and used a Thermo FlashEA® 1112 elemental analyser.
Results and discussion
Mesoporous silica powders and EASA films were produced with hexagonal pore structure (space group P6mm) and a similar lattice spacing. This allowed the same grafting procedures to be applied to both, providing access to a broad range of analytical techniques that can only be applied to powder samples and allowing comparisons between the two systems.Mesoporous silica powders
Hexagonal mesoporous silica was produced from TEOS and CTAB using an adaptation of a published route.4 SAXS measurements on these samples (Fig. 1) showed the 10 (2.28°), 11 (3.92°) and 20 (4.52°) reflections of hexagonal mesoporous silica with a pore spacing equal to the lattice parameter of a = 4.50(1) nm. The porosity of the unmodified mesoporous silica powders was analysed using nitrogen sorption (Fig. 2). The isotherms adopted the IUPAC type IV profile that is typical for these MCM-41 type materials, with similar amounts of monolayer adsorption on the silica surfaces (low pressure) and pore filling (P/P0 = 0.2–0.4) as reported previously.4 The BET analysis measured the surface area as being 1093 m2 g−1 and the pore size distribution calculated using NLDFT was centred on 2.95 nm (Fig. 2). Classically the analysis of pore sizes in gas sorption data used the Barrett–Joyner–Halenda (BJH) method, but it has been shown this approach underestimates pore sizes in small mesopores and in the microporous regimes.47 The NLDFT approach used herein was developed by Seaton et al.,44 received ISO standardisation in 2007,43 and has been found to be reliable when used on MCM-41 silicas.48–50 | ||
| Fig. 1 The SAXS pattern of mesoporous silica powder and the in-plane GISAXS patterns of mesoporous silica films deposited at conditions as labelled. | ||
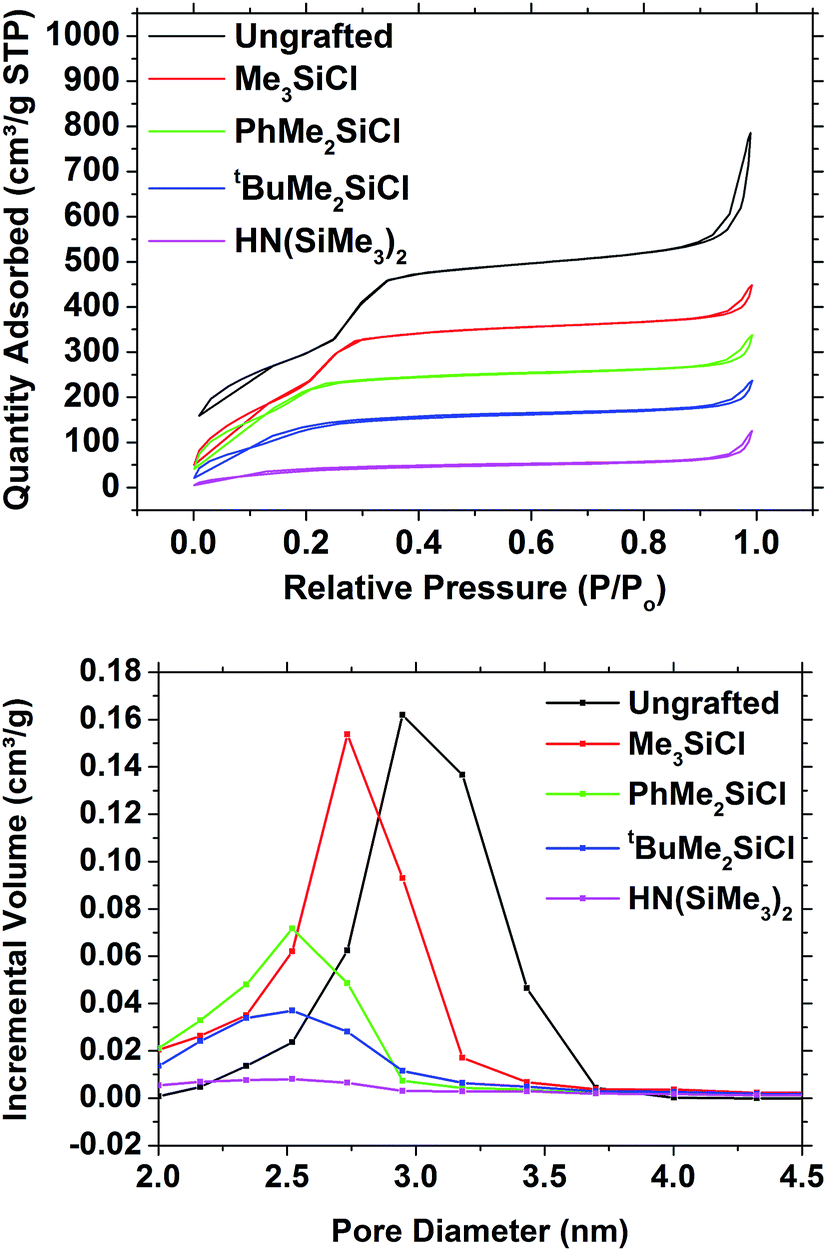 | ||
| Fig. 2 Nitrogen adsorption/desorption isotherms (top) and NLDFT pore size distribution (bottom) of mesoporous silica powder and of grafted mesoporous silica powders. | ||
EASA mesoporous silica films with vertically aligned pores
We previously reported the EASA deposition of mesoporous silica films on PVD TiN films.27 Herein the substrate type was changed to a commercial MOCVD TiN with higher electronic conductivity, and this affected the deposition conditions under which the EASA films could be deposited to a surprising degree. Depositions under different conditions chosen for roughly the same amount of charge to have been passed resulted in the GISAXS patterns shown in Fig. 1 and analysed in Table 1. The 10, 11, 20 and 21 reflections associated with a hexagonal (P6mm) pore structure were clearly visible in the in-plane GISAXS, whereas only very broad, weak features close to the position of the 10 reflection were observed out-of-plane. This confirms that the pores are vertically aligned with negligible horizontal alignment. The pore spacing of 4.36(2) nm observed in these films is also similar to that observed in our previous work,27 where the vertical pore structure was also extensively characterised by electron microscopy, and in the powdered silica (4.50(3) nm). The most intense GISAXS pattern was obtained after a deposition held at −1.3 V for 20 s, so these conditions were chosen to produce the samples used in subsequent studies. An exemplar TEM image of one of these films, confirming the pore orientation, is shown in Fig. S1.†| Deposition conditions | 2θ(10)/° | 2θ(11)/° | 2θ(20)/° | 2θ(21)/° | a/nm |
|---|---|---|---|---|---|
| −1.8 V, 5 s | 2.28 | 3.98 | 4.62 | — | 4.44(2) |
| −1.6 V, 10 s | 2.34 | 4.04 | 4.68 | 6.20 | 4.364(7) |
| −1.3 V, 20 s | 2.34 | 4.06 | 4.70 | 6.18 | 4.351(7) |
Ellipsometric porosimetry can be used to provide information on the pore structure of thin films,45,51 performing the same role as nitrogen adsorption/desorption measurements with powders. These measurements are performed with a sample placed in a sealed chamber and a laser is reflected off the surface of the thin-film sample. The initial measurements are taken under vacuum conditions and the refractive index of the film is determined. The humidity in the chamber is then increased and the refractive index is continually measured. As the film is exposed to vapour of a probe molecule such as water or toluene, condensation occurs within the porous structure of the film and causes a change in the refractive index of the film. By monitoring the change in the refractive index of the sample as the partial pressure is increased, the absorption/desorption isotherm can be determined.
The pore structure of the EASA silica films was analysed by ellipsometric porosimetry using water, toluene, isopropyl alcohol and methanol probe molecules. All four probe molecules produced type IV isotherms (Fig. 3), with the partial pressures at which capillary condensation occurs varying roughly with the polarity of the probe molecule. Toluene wets the pores and condenses at low pressure, but desorbs cleanly over the same pressure range, returning to its original refractive index at low pressure. The alcohol and water isotherms exhibit hysteresis due to hydrogen bonding to the pore wall hydroxyl groups. Unsurprisingly this effect is strongest with water, where incomplete removal of the water by the vacuum is also apparent from a higher final refractive index at the end of the desorption process than at the start of the experiment. A large hysteresis was previously observed in ellipsometric porosimetry of EASA films on ITO using water vapour.52 Porosities of the EASA films calculated from the adsorption branches of these isotherms were 38.2% using toluene, 40.1% using water, 42.6% using isopropyl alcohol and 42.5% using methanol. These are in good mutual agreement, although clearly some variability between films is a clear possibility. Powders produced with a CTAB surfactant have pore sizes in the range 3.0–5.7 Å, depending on a range of factors during the synthesis.4,53–55 The electrochemical EASA growth method results in smaller pores, with 1.6 nm pores found in EASA silica films on TiN.27 A 4.46 nm pore spacing combined with this pore size would result in a porosity of 31%.
 | ||
| Fig. 3 Plots of refractive index (n) of an EASA mesoporous silica film vs. solvent vapour partial pressure during adsorption and desorption of toluene, water, isopropyl alcohol or methanol. | ||
Analysis of surface area and pore size was considered to be most effective with the least polar solvents, where both the adsorption and desorption branches could be used. It has been argued that porous confinement can distort results using a polar solvent due to the greater degree of polarisation present in the surface/adsorbate interaction.56 The BET surface area of the film was determined to be 1093 m2 g−1 using the toluene isotherm. Application of a modified Kelvin equation45 to the toluene isotherm led to a pore diameter of 2.72 nm using the adsorption branch or 2.74 nm using the desorption. This is higher than previously observed in EASA silica27 but it has been previously acknowledged that the Kelvin equation ignores intermolecular interactions that become increasingly important at small pore size, and hence is likely to overestimate the pressure required to wet pores in the micropore regime (<2 nm (ref. 46)). The Dubinin–Rasushkevich (DR) model assumes that liquid in the pore channels is homogeneously filled and has liquid-like properties, and hence becomes more accurate as the intermolecular interactions become dominant in the micropore region.57 Note that the samples studied herein were expected to have a pore size at the upper end of this regime. Using the DR model with the toluene isotherm (Fig. 4) the pore size was determined to be 1.44 nm (adsorption branch) or 1.48 nm (desorption branch). Using the isopropyl alcohol isotherms the values were slightly smaller, at 1.14 nm (adsorption) or 1.20 nm (desorption), reflecting the larger wall interaction. Hence the toluene data fitted with the DR model were found to provide the most plausible pore size results.
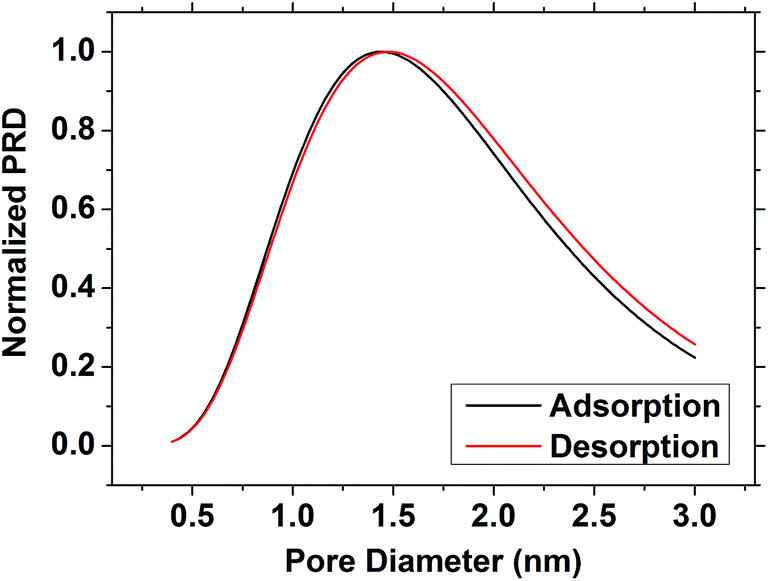 | ||
| Fig. 4 The pore size distribution of the EASA silica film as determined using the toluene isotherm and the Dubinin–Radushkevich model. | ||
Surface modification of mesoporous silica powders
Surface treatments of mesoporous silica utilized trialkyl (/aryl) chlorosilanes or hexamethyldisilazane. The dried powder silica samples were refluxed with these compounds in THF. They were then subjected to a thorough washing regime to remove as much of the unreacted grafting agent as possible, including a Soxhlet extraction. The SAXS patterns were essentially indistinguishable from those of the ungrafted silica powder (Fig. S2†), so the mesoporous structure underwent no significant changes in the pore ordering. The in-plane GISAXS measurements gave lattice parameters of 4.493(12) nm for ungrafted powder, 4.441(8) nm for Me3SiCl grafted powder, 4.312(8) nm for PhMe2SiCl grafted powder, 4.36(2) nm for tBuMe2SiCl grafted powder and 4.231(14) nm for HN(SiMe3)2 grafted powders.The nitrogen adsorption–desorption isotherms (Fig. 2) showed a gradual decrease in the size of the capillary condensation feature as the size of the grafting group increased, and at the same time a reduction both in the pore diameter and the pore volume. The surface area of the samples decreased from the 1093 m2 g−1 in the ungrafted silica to 904 m2 g−1 with Me3SiCl, 729 m2 g−1 with PhMe2SiCl, 538 m2 g−1 with tBuMe2SiCl and 161 m2 g−1 with HN(SiMe3)2. The NLDFT pore diameter decreased from 2.95 nm to 2.73, 2.52, 2.52 and 2.50 nm in the same sequence, although a significant loss of pore volume was also observed with the larger agents (Fig. 2), suggesting some pore blocking was also occurring.
The 29Si MAS-NMR spectrum of the ungrafted silica powder (Fig. 5) contained two broad signals at −98.7 and −107.5 ppm, corresponding to the Q3 (Si(OH)(OSi)3) and Q4 (Si(OSi)4) environments within the silica.58,59 These persisted after grafting (with peak shifts of no more than 1.5 ppm between samples), but were then also accompanied by signals at in the range of 3 to 16 ppm (14.1, 3.6, 16.7 and 12.8 respectively for Me3SiCl, PhMe2SiCl, tBuMe2SiCl and HN(SiMe3)2) corresponding to the grafted silane molecules.59 The upfield shift for the Ph-substituted silane and downfield shift for the tBu substituted silane are in line with trends observed in the literature.60 These are significantly different from the 29Si chemical shifts of the chlorosilane reagents, provided in Blinka et al.60 for Me3SiCl, tBuMe2SiCl and HN(SiMe3)2, suggesting that no unreacted materials are present in the systems studied here. The strongest signals are present in the Me3SiCl and PhMe2SiCl grafted powders, suggesting that these samples contained the largest number of grafted sites. As the Si(OH)(OSi)3 sites are reacted with the grafting agent the Q3 signal in the grafted samples should decrease relative to Q4 and to the Q3 signal in the ungrafted membrane.61 The CP spectra (Fig. 5) are not quantitative so direct acquisition spectra were also collected (Fig. S3†) and these confirm the expected decrease in the Q3 signal.
 | ||
| Fig. 5 29Si (top) and 13C (bottom) CP MAS-NMR spectra of the mesoporous silica powder samples. | ||
The 13C spectra (Fig. 5) show the presence of some surfactant impurities (signals near 54 ppm for NMe3 group and signals between 20–35 ppm for the long aliphatic chain of CTAB). This indicates that there is still CTAB present in several of the films, but also that the Me3SiCl grafting removed almost all of the remaining CTAB from the film. Both 29Si and 13C NMR data suggest that there is no tetraethoxysilane left as unreacted material in the sample. The 13C signal for Si–Me groups is assigned to the broad signal near 0 ppm60 and it is a clear signature for the successful grafting on the surface. Interestingly, the 13C data correlate well with the 29Si data to indicate that in samples with good grafting levels as detected by 29Si NMR (Me3SiCl and PhMe2SiCl), the corresponding signals from the grafted species are clearly visible by 13C NMR. The PhMe2SiCl species give additional signals near 130–140 ppm. For the other samples (tBuMe2SiCl and HN(SiMe3)2) where there is a very weak signal from grafted silicon via29Si NMR and the 13C signal from Si–Me is also very weak. Both 13C and 29Si data suggests that free starting material for the grafting process which did not bond were effectively removed, and do not appear in the 13C or 29Si cross-polarisation spectra.
Combustion analysis showed the ungrafted mesoporous silica powder to contain significant carbon (Table 2). The C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N ratio of CTAB is 19:1, similar to the analysed value, so the surfactant seen in the NMR data is likely to be the main contributor. The TEOS starting material or the ethanol used in the Soxhlet extraction of the surfactant could also contribute. Mesoporous silica syntheses often involve firing to burn away the surfactant, but oxidation of the TiN surface of the films precludes this approach. The grafted samples (Table 2) show lower carbon contents and a higher C:N ratio, suggesting the surfactant is displaced from the pore during grafting. It is possible that the Soxhlet extraction failed to completely remove the surfactant due to [CTA]+ cations balancing surface SiO− groups, but that these react with the grafting agent releasing the surfactant. Me3SiCl and PhMe2SiCl show the greatest change in the analysis and were most effective at removing the surfactant, as also shown by the 13C NMR data (Fig. 5). The analysis of the sample grafted with HN(SiMe3)2 was close to that of the ungrafted film, indicating that this grafting agent was least successful in releasing surfactant from the pores. The carbon content of Me3SiCl and PhMe2SiCl grafted films is mainly due to the grafting agent, and taking the number of carbon atoms per grafting molecule into account the coverage of Me3Si groups (11.3/3 = 3.8) is seen to be higher than that of PhMe2Si groups (15.8/8 = 1.9).
:N ratio of CTAB is 19:1, similar to the analysed value, so the surfactant seen in the NMR data is likely to be the main contributor. The TEOS starting material or the ethanol used in the Soxhlet extraction of the surfactant could also contribute. Mesoporous silica syntheses often involve firing to burn away the surfactant, but oxidation of the TiN surface of the films precludes this approach. The grafted samples (Table 2) show lower carbon contents and a higher C:N ratio, suggesting the surfactant is displaced from the pore during grafting. It is possible that the Soxhlet extraction failed to completely remove the surfactant due to [CTA]+ cations balancing surface SiO− groups, but that these react with the grafting agent releasing the surfactant. Me3SiCl and PhMe2SiCl show the greatest change in the analysis and were most effective at removing the surfactant, as also shown by the 13C NMR data (Fig. 5). The analysis of the sample grafted with HN(SiMe3)2 was close to that of the ungrafted film, indicating that this grafting agent was least successful in releasing surfactant from the pores. The carbon content of Me3SiCl and PhMe2SiCl grafted films is mainly due to the grafting agent, and taking the number of carbon atoms per grafting molecule into account the coverage of Me3Si groups (11.3/3 = 3.8) is seen to be higher than that of PhMe2Si groups (15.8/8 = 1.9).
| Grafting agents | Carbon (%) | Hydrogen (%) | Nitrogen (%) | Ratio of C/N |
|---|---|---|---|---|
| Ungrafted | 26.8 | 5.4 | 1.5 | 17.7 |
| Me3SiCl | 11.3 | 2.7 | <0.1 | 112.7 |
| PhMe2SiCl | 15.4 | 2.6 | 0.2 | 66.9 |
| t BuMe2SiCl | 18.7 | 3.9 | 0.9 | 20.3 |
| HN(SiMe3)2 | 24.8 | 5.0 | 1.3 | 19.0 |
Combined, the NMR and analytical data suggest significant retention of CTAB in the Soxhlet extracted powders. Smaller grafting agents can release this from the pores during the grafting process. The CTAB was not blocking pores significantly according to the ungrafted powder porosimetry, but some reorganisation of the CTAB may result in it blocking pores in conjunction with the larger grafting agents. For example, action of these agents on particle surfaces and pore entrances could result in production of HCl which could migrate into the pores to release CTA+ ions as the chloride salt and protonate the silica. The smaller grafting agents may react simultaneously at sites deeper in the pores and at the surfaces simultaneously, releasing the surfactant and avoiding this effect.
Surface modification of mesoporous silica films
EASA films were grafted with the same silane reagents as the powders and under the same reaction conditions. In-plane GISAXS patterns were very similar to those of the ungrafted films both before and after Soxhlet extraction in acidified ethanol (Fig. S4†). The lattice parameters extracted from these patterns were 4.35(8) nm for an ungrafted silica film, 4.345(16) nm for a Me3SiCl grafted film, 4.39(3) nm for a PhMe2SiCl grafted film and 4.396(18) nm for a tBuMe2SiCl grafted film. The lattice parameters after the films were Soxhlet extracted were unchanged at 4.369(19) nm (Me3Si), 4.345(16) nm (PhMe2SiCl) and 4.384(18) nm (tBuMe2SiCl).Water contact angle measurements were used to determine the surface hydrophobicity of the films. The ungrafted film was fairly hydrophilic, with the water droplet spreading significantly (Fig. 6) and making a contact angle of 61°. The grafted films were all significantly more hydrophobic and the response varied largely with the hydrophobicity of the grafting group, from 84° with PhMe2SiCl, to 89° or 90°, respectively, when grafted with Me3SiCl or HN(SiMe3)2, and 97° when grafted with tBuMe2SiCl (Fig. 6). The TiN substrate had a contact angle of 79°. This shows that all four grafting agents were effective at modifying the top surface of the films, but does not give an indication of their effectiveness at modifying the pore walls. Hence ellipsometric porosimetry was also employed to probe this. The presence of CTAB in the pores of several of the films, as indicated by the 13C NMR and the combustion analysis, is a potential cause of the hydrophobic character of the surface of the membranes. However the degree of hydrophobicity is smallest in the ungrafted membrane, which contains the highest amount of CTAB. The degree of hydrophobicity is also greater in the Me3SiCl grafted membrane, where the grafting agent removed all of the CTAB from the pores. This shows that the CTAB has little effect on the surface hydrophobicity of the films.
 | ||
| Fig. 6 Images of 1 μL water droplets on EASA silica films ungrafted (left) and grafted with Me3SiCl (right) for the purpose of contact angle measurement. | ||
Since the HN(SiMe3)2 had been found ineffective at grafting the surfaces of the larger pores in the powder samples, the ellipsometric porosimetry studies on the thin films concentrated on the three chlorosilane reagents. With toluene the three films all exhibited the same basic shape of isotherm as found in the ungrafted film (Fig. 7). This is unsurprising since toluene wetted the silica films and would also be expected to wet the more hydrophobic grafted films. The porosity (Table 3) was measured with toluene as 30.4% with Me3SiCl, 29% with the larger PhMe2SiCl and 37.3% with tBuMe2SiCl. This final value was closer to the 38.2% measured on an ungrafted film. Interestingly the isotherm with PhMe2SiCl exhibited two clear features in the capillary evaporation region of the desorption isotherm and this feature was even more pronounced in the methanol and isopropanol isotherms (Fig. S5–S7†), suggesting the presence of two pore diameters. The lack of this feature in the adsorption isotherms suggests that these are not separate pores, but that the top part of the pores is narrowed due to grafting only the most accessible region. The bottom regions of the pores may be less accessible to the larger reagents. Grafting at pore entrances has also been observed in silica powders.62–64 Pore diameters measured with toluene (Table 3) were found to increase after grafting, consistent with surfactant removal as seen in the powders.
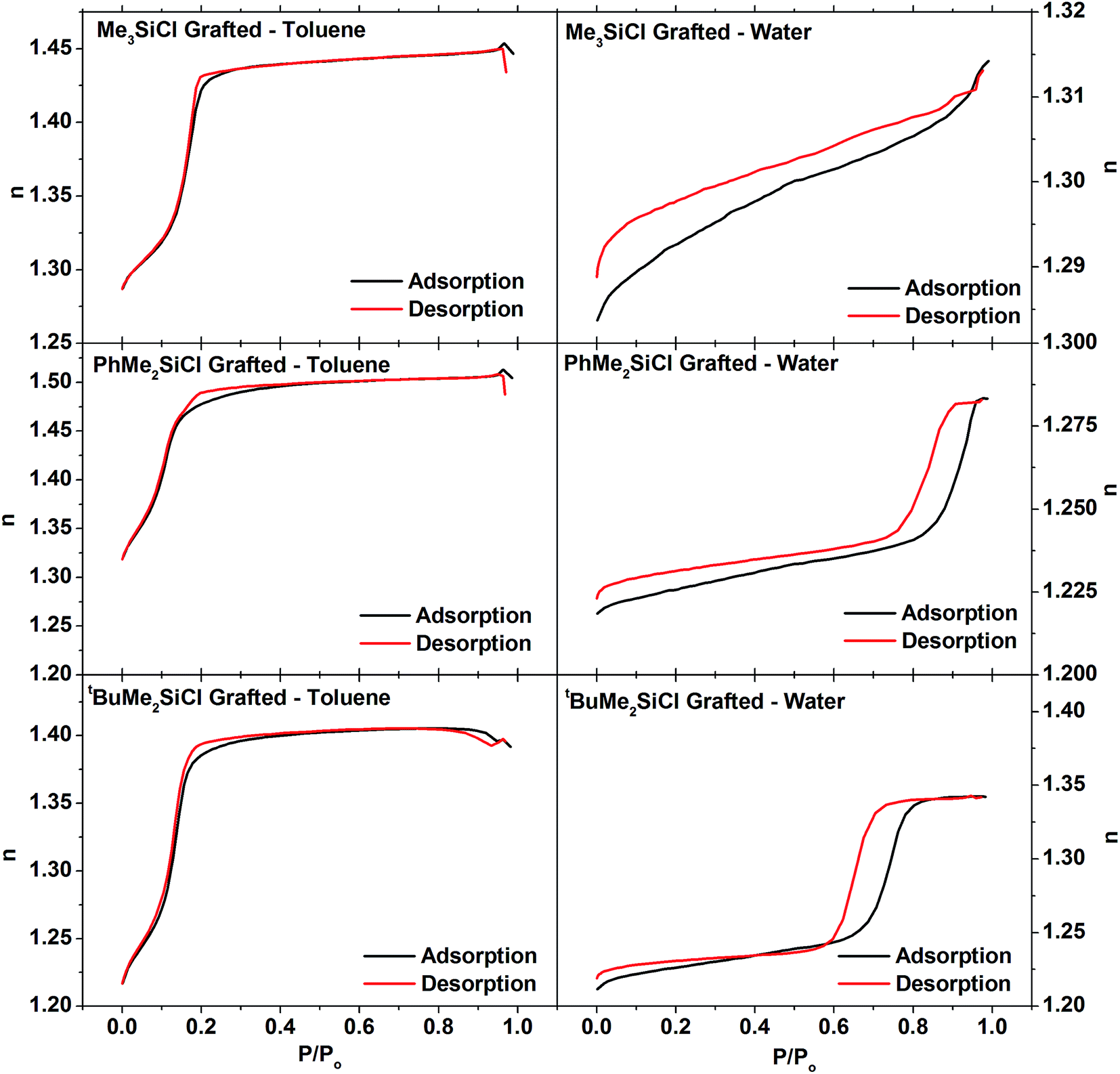 | ||
| Fig. 7 Ellipsometric porosimetry isotherms measured with toluene (left) or water (right) of EASA silica films grafted with Me3SiCl (top), PhMe2SiCl (middle) or tBuMe2SiCl (bottom). | ||
| Grafting agent | Solvent | Porosity (%) | Mod. Kelvin eqn diameter (nm) | DR diameter (nm) |
|---|---|---|---|---|
| Ungrafted | Toluene | 38.2 | 2.72abs/2.74des | 1.44abs/1.48des |
| Water | 40.1 | — | — | |
| IPA | 42.6 | 2.74abs/2.68des | 1.14abs/1.20des | |
| Methanol | 42.5 | — | — | |
| Me3SiCl | Toluene | 30.4 | 3.14abs/3.14des | 1.54abs/1.52des |
| Water | 7.7 | — | — | |
| IPA | 30.7 | 2.86abs/2.84des | 1.30abs/1.36des | |
| Methanol | 29.26 | — | — | |
| PhMe2SiCl | Toluene | 29.0 | 2.50ads/2.44des | 1.68abs/1.76des |
| Water | 18.8 | — | — | |
| IPA | 28.4 | 2.32ads/2.30des | 1.36abs/1.38des | |
| Methanol | 27.6 | — | — | |
| t BuMe2SiCl | Toluene | 37.3 | 2.76ads/2.66des | 1.50ads/1.54des |
| Water | 35.8 | — | — | |
| IPA | 38.7 | 2.66ads/2.62des | 1.02ads/1.12des | |
| Methanol | 36.8 | — | — |
The isotherm of the Me3SiCl-grafted film with water shows virtually no capillary condensation and only a very small volume of absorbed vapour at pressures beneath the bulk water vapour pressure. Reconciling these results with the toluene results on the same film requires that a significant fraction, ≈75%, of the pores within the treated film are now hydrophobic. The PhMe2SiCl grafted film also shows a reduction in pore fraction accessible to water at pressure beneath the bulk water vapour pressure although in this case only ≈40% of the pores are hydrophobic. On the other hand, the tBuMe2SiCl treated films show only a very small difference in the porosities measured with water and the other three solvents which suggests very little change in the hydrophilicity of the pores in this film. This is consistent with the poor coverage and surfactant removal observed with this grafting agent in powders. The results also help to explain variable effects on the behaviour of hydrophilic redox probes in EASA films with different surface treatments.37,39
Overall it is clear that the grafting process fundamentally preserves the pore structure of the mesoporous film for all of the grafting agents as all of the grafted films show significant porosity when measured with toluene. The smallest grafting agent, Me3SiCl, shows the clearest evidence of significant pore modification, with the porosity measured using water being only 25% of the porosity measured using toluene. PhMe2SiCl, which is slightly larger, also causes a significant change in the hydrophilicity of the pores however to a lesser extent than Me3SiCl; water porosity is 60% of the toluene porosity. In the case of tBuMe2SiCl there is very little evidence that it has modified the pores of the film. In particular, the pore volume fraction measured with this film (37%) is to within error the same as for an untreated film (38%) whereas the other two treated films have pore volume fractions nearer 30%. In addition this film, like untreated films, shows no difference in the porosity measured with water and the other three solvents. Whilst tBuMe2SiCl was able to penetrate the pores of the powered mesoporous silica to some degree, it is clear that the pores in the films are significantly smaller (∼1.6 nm) compared to the powered samples (∼3 nm).
The presence of residual CTAB in the pores after grafting, as shown by the NMR and combustion analysis, could potentially contribute to the hydrophobic character of the pores. However the porosimetry measurements show that this is not the case. The porosity measured with water in the ungrafted film, which contains the most CTAB, is 40.1%, while the equivalent porosity measurement in the Me3SiCl grafted film, the film which contains almost no CTAB, shows 7.7%. This is also shown for the other grafted films having greater hydrophobicity than the ungrafted film. This indicates that the presence of CTAB has no influence on the hydrophobic character of the films.
All of the evidence taken together suggests that for pores in the films the diameter that the size of the grating agent is critical to the successful grating of the pores. This constitutes a novel route to control pore sizes in vertically aligned hexagonal mesoporous silica films, opening new possibilities for fine tuning of the molecular sieving properties of such oriented membranes,65 and to control pore wall chemistry with the possibility of allowing ingress of reagents or solvents for electrodeposition.
Conclusions
Hexagonal mesoporous silica powder and vertically aligned films have been synthesized using TEOS and CTAB surfactant. Both exhibited good hexagonal order in the pore structures and similar pore spacings. The nitrogen sorption measurements showed a surface area of approximately 1100 m2 g−1 for the powder. The ellipsometric porosimetry measurements gave a pore diameter of approximately 1.4 nm using the Dubinin–Rasushkevich model and 2.7 nm using a modified Kelvin equation. The former was consistent with previous TEM evidence.The mesoporous silica powders and films were successfully grafted with various silane reagents. The SAXS measurements showed little difference in the pore structure between the ungrafted and grafted mesoporous films and powders. The 29Si NMR analysis showed that the grafted powders contained extra Si environments corresponding to the silane groups grafted onto the pore walls. The CHN analysis indicated that some CTAB surfactant was still present in the pores of the ungrafted silica powders, but was displaced by the grafting with silane groups. DFT calculations showed a decrease in the pore diameter between the ungrafted and the grafted powders. A general trend was observed for the different silane grafting reagents that the reagents with the smallest steric bulk gave the greater coverage of the pore walls and largest grafted species peaks in the 29Si NMR spectra. This indicates that the bulkier reagents are unable to fully penetrate the pore and fully graft the inside of the pore wall.
Ellipsometric porosimetry of the grafted mesoporous silica films showed that the penetration of water greatly decreased when grafted with the smaller silane reagents such as Me3SiCl, which was clearly effective in making the pore walls hydrophobic. However there was no change when grafting with the larger silane reagents such as tBuMe2SiCl, and evidence for partial grafting at the top of the pores with PhMe2SiCl, further indicating that the size of the grafting agent affects the degree of grafting undertaken in the pore structure. Surfactant persisted in the pores after Soxhlet extraction, but this did not significantly affect hydrophobicity. The smaller pores in the silica films relative to the powders resulted in a higher degree of exclusion of the larger grafting agents.
Acknowledgements
The authors thank EPSRC for funding under the Supercritical Fluids Electrodeposition project (SCFED, EP/I033394/1) and for funding the Smartlab diffractometer (EP/K00509X/1 and EP/K009877/1). The SCFED Project (www.scfed.net) is a multidisciplinary collaboration of British universities investigating the fundamental and applied aspects of supercritical fluids. Marina Carravetta thanks the Royal Society for a University Research Fellowship.References
- I. K. Mbaraka and B. H. Shanks, J. Catal., 2005, 229, 365–373 CrossRef CAS.
- A. Walcarius, Chem. Soc. Rev., 2013, 42, 4098–4140 RSC.
- B. J. Melde, B. J. Johnson and P. T. Charles, Sensors, 2008, 8, 5202–5228 CrossRef CAS PubMed.
- R. I. Nooney, D. Thirunavukkarasu, Y. Chen, R. Josephs and A. E. Ostafin, Chem. Mater., 2002, 14, 4721–4728 CrossRef CAS.
- T. Maschmeyer, F. Rey, G. Sankar and J. M. Thomas, Nature, 1995, 378, 159–162 CrossRef CAS.
- J. Zhang, Z. Ma, J. Jiao, H. Yin, W. Yan, E. W. Hagaman, J. Yu and S. Dai, Microporous Mesoporous Mater., 2010, 129, 200–209 CrossRef CAS.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- D. J. Macquarrie, Green Chem., 1999, 195–198 RSC.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- R. Luque, A. M. Balu, J. M. Campelo, M. D. Gracia, E. Losada, A. A. R. Pineda and J. C. Serrano-Ruiz, in Catalysis: Volume 24, ed. J. J. Spivey and M. Gupta, RSC Publishing, Cambridge, UK, 2012, pp. 253–280 Search PubMed.
- N. Lashgari, A. Badiei and G. M. Ziarani, Nanochemistry Research, 2016, 1, 127–141 Search PubMed.
- Y. G. Jin, S. Z. Qiao, Z. P. Xu, Z. Yan, Y. Huang, J. C. Diniz da Costa and G. Q. Lu, J. Mater. Chem., 2009, 19, 2363 RSC.
- A. Walcarius, Electroanalysis, 2015, 27, 1303–1340 CrossRef CAS.
- M. Etienne, L. Zhang, N. Vila and A. Walcarius, Electroanalysis, 2015, 27, 2028–2054 CrossRef CAS.
- L. Bois, A. Bonhommé, A. Ribes, B. Pais, G. Raffin and F. Tessier, Colloids Surf., A, 2003, 221, 221–230 CrossRef CAS.
- A. Walcarius and L. Mercier, J. Mater. Chem., 2010, 20, 4478 RSC.
- A. Nomura and C. W. Jones, ACS Appl. Mater. Interfaces, 2013, 5, 5569–5577 CAS.
- I. Andreou, H. Amenitsch, V. Likodimos, P. Falaras, P. Koutsoukos and E. Leontidis, Materials, 2013, 6, 1467–1484 CrossRef CAS.
- Y.-F. Lee, K.-H. Chang, C.-Y. Chu, H.-L. Chen and C.-C. Hu, RSC Adv., 2011, 1, 401–407 RSC.
- D. Grosso, F. Cagnol, G. J. de A. A. Soler-Illia, E. L. Crepaldi, H. Amenitsch, A. Brunet-Bruneau, A. Bourgeois and C. Sanchez, Adv. Funct. Mater., 2004, 14, 309–322 CrossRef CAS.
- P. Innocenzi and L. Malfatti, Chem. Soc. Rev., 2013, 42, 4198–4216 RSC.
- A. Walcarius, E. Sibottier, M. Etienne and J. Ghanbaja, Nat. Mater., 2007, 6, 602–608 CrossRef CAS PubMed.
- A. Walcarius and A. Kuhn, TrAC, Trends Anal. Chem., 2008, 27, 593–603 CrossRef CAS.
- M. Etienne, A. Goux, E. Sibottier and A. Walcarius, J. Nanosci. Nanotechnol., 2009, 9, 2398–2406 CrossRef CAS PubMed.
- Y. Guillemin, J. Ghanbaja, E. Aubert, M. Etienne and A. Walcarius, Chem. Mater., 2014, 26, 1848–1858 CrossRef CAS.
- G. Herzog, E. Sibottier, M. Etienne and A. Walcarius, Faraday Discuss., 2013, 164, 259–273 RSC.
- C. Robertson, R. Beanland, S. A. Boden, A. L. Hector, R. J. Kashtiban, J. Sloan, D. C. Smith and A. Walcarius, Phys. Chem. Chem. Phys., 2015, 17, 4763–4770 RSC.
- Z. Teng, G. Zheng, Y. Dou, W. Li, C.-Y. Mou, X. Zhang, A. M. Asiri and D. Zhao, Angew. Chem., Int. Ed. Engl., 2012, 51, 2173–2177 CrossRef CAS PubMed.
- M. Hara, S. Nagano and T. Seki, J. Am. Chem. Soc., 2010, 132, 13654–13656 CrossRef CAS PubMed.
- Y. Guillemin, M. Etienne, E. Aubert and A. Walcarius, J. Mater. Chem., 2010, 20, 6799–6807 RSC.
- A. Goux, M. Etienne, E. Aubert, C. Lecomte, J. Ghanbaja and A. Walcarius, Chem. Mater., 2009, 21, 731–741 CrossRef CAS.
- E. M. Björk, Mesoporous Building Blocks – Synthesis and Characterization of Mesoporous Silica Particles and Films, PhD thesis, Linkoping University, 2013.
- D. J. Macquarrie, Top. Catal., 2009, 52, 1640–1650 CrossRef CAS.
- P. N. Bartlett, J. Burt, D. A. Cook, C. Y. Cummings, M. W. George, A. L. Hector, M. M. Hasan, J. Ke, W. Levason, D. Pugh, G. Reid, P. W. Richardson, D. C. Smith, J. Spencer, N. Suleiman and W. Zhang, Chem.–Eur. J., 2015, 302–309 Search PubMed.
- P. N. Bartlett, C. Y. Cummings, W. Levason, D. Pugh and G. Reid, Chem.–Eur. J., 2014, 20, 5019–5027 CrossRef CAS PubMed.
- C. Y. Cummings, P. N. Bartlett, D. Pugh, G. Reid, W. Levason, M. M. Hasan, A. L. Hector, J. Spencer and D. C. Smith, J. Electrochem. Soc., 2015, 162, D619–D624 CrossRef CAS.
- N. Vilà, J. Ghanbaja, E. Aubert and A. Walcarius, Angew. Chem., Int. Ed. Engl., 2014, 53, 2945–2950 CrossRef PubMed.
- N. Vilà, J. Ghanbaja and A. Walcarius, Adv. Mater. Interfaces, 2016, 3, 1500440 CrossRef.
- M. Rafiee, B. Karimi, S. Farrokhzadeh and H. Vali, Electrochim. Acta, 2013, 94, 198–205 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- J. Landers, G. Y. Gor and A. V. Neimark, Colloids Surf., A, 2013, 437, 3–32 CrossRef CAS.
- A. M. Puziy, O. I. Poddubnaya, B. Gawdzik and M. Sobiesiak, Adsorption, 2016, 22, 459–464 CrossRef CAS.
- ISO 15901-32007, Pore size Distrib. porosity solid Mater. by Mercur. Porosim. gas Adsorpt. 3 Anal. micropores by gas Adsorpt, https//www.iso.org/obp/ui/#isostdiso15901-3ed-1v1en,.
- N. A. Seaton, J. P. R. B. Walton and N. Quirke, Carbon, 1989, 27, 853–861 CrossRef CAS.
- M. R. Baklanov, K. P. Mogilnikov, V. G. Polovinkin and F. N. Dultsev, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2000, 18, 1385 CrossRef CAS.
- C. Lastoskie, K. E. Gubbins and N. Quirke, J. Phys. Chem., 1993, 97, 4786–4796 CrossRef CAS.
- M. Luisa Ojeda, J. Marcos Esparza, A. Campero, S. Cordero, I. Kornhauser and F. Rojas, Phys. Chem. Chem. Phys., 2003, 5, 1859 RSC.
- A. V. Neimark and P. I. Ravikovitch, Microporous Mesoporous Mater., 2001, 44, 697–707 CrossRef.
- M. Thommes, B. Smarsly, M. Groenewolt, P. I. Ravikovitch and A. V. Neimark, Langmuir, 2006, 22, 756–764 CrossRef CAS PubMed.
- P. I. Ravikovitch, a. Vishnyakov and a. V. Neimark, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2001, 64, 011602 CrossRef CAS PubMed.
- A. Bourgeois, Y. Turcant, C. Walsh and C. Defranoux, Adsorption, 2008, 14, 457–465 CrossRef CAS.
- M. Etienne, Y. Guillemin, D. Grosso and A. Walcarius, Anal. Bioanal. Chem., 2013, 405, 1497–1512 CrossRef CAS PubMed.
- A. Wang and T. Kabe, Chem. Commun., 1999, 2067–2068 RSC.
- M. Gru, K. K. Unger, A. Matsumoto and K. Tsutsumi, Microporous Mesoporous Mater., 1999, 27, 207–216 CrossRef.
- T. Asefa and Z. Tao, Can. J. Chem., 2012, 90, 1015–1031 CrossRef CAS.
- C. Boissiere, D. Grosso, S. Lepoutre, L. Nicole, A. B. Bruneau and C. Sanchez, Langmuir, 2005, 21, 12362–12371 CrossRef CAS PubMed.
- M. Thommes, Chem.-Ing.-Tech., 2010, 82, 1059–1073 CrossRef CAS.
- R. Simonutti, A. Comotti, S. Bracco and P. Sozzani, Chem. Mater., 2001, 13, 771–777 CrossRef CAS.
- P. Sutra, F. Fajula, D. Brunel, P. Lentz, G. Daelen and J. B. Nagy, Colloids Surf., A, 1999, 158, 21–27 CrossRef CAS.
- T. A. Blinka, B. J. Helmer and R. West, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Academic Press Inc., New York, 1984, pp. 193–218 Search PubMed.
- C. Henrist, C. Vogels, A. Rulmont and R. Cloots, New J. Chem., 2005, 29, 1017–1021 RSC.
- M. H. Lim and A. Stein, Chem. Mater., 1999, 11, 3285–3295 CrossRef CAS.
- L. Mercier and T. J. Pinnavaia, Chem. Mater., 2000, 12, 188–196 CrossRef CAS.
- R. J. P. Corriu, E. Lancelle-Beltran, A. Mehdi, C. Reyé, S. Brandès and R. Guilard, Chem. Mater., 2003, 15, 3152–3160 CrossRef CAS.
- N. Vilà, E. André, R. Ciganda, J. Ruiz, D. Astruc and A. Walcarius, Chem. Mater., 2016, 28, 2511–2514 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra23059h |
| ‡ Raw data are also available from DOI: 10.5258/SOTON/402889. |
| This journal is © The Royal Society of Chemistry 2016 |