
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterization of the laccase-mediated oligomerization of 4-hydroxybenzoic acid†
Sjoerd
Slagman
,
Jorge
Escorihuela
,
Han
Zuilhof
and
Maurice C. R.
Franssen
 *
*
Laboratory of Organic Chemistry, Wageningen University, Stippeneng 4, building 124 (Helix), 6708 WE Wageningen, The Netherlands. E-mail: Maurice.franssen@wur.nl; Web: www.orc.wur.nl
First published on 12th October 2016
Abstract
Modifying inert poly(ethersulfone) membranes using laccase has proven to be an environmentally benign and easily applicable process to alter the membrane's surface properties. By this method phenolic acid monomers such as 4-hydroxybenzoic acid are grafted from the membrane surface to make it anti-fouling. In order to enhance the anti-fouling capabilities even further it is important to study the molecular details of this reaction. However, the nature of the products of laccase modification, either on a surface or in solution, has been studied only poorly. In this paper we report the formation of C3–C3′, C3–O and C1–C3′ linked dimers as the first products of the solution-phase laccase-mediated oligomerization of 4-hydroxybenzoic acid. These dimers can also act as substrate for laccase, and we show that their enzymatic oxidative coupling occurs far more rapidly than that of 4-hydroxybenzoic acid, which indicates that they are highly reactive intermediates that are efficiently polymerized onwards. The reactivity of each dimer is of large influence on its yield; dimers that are converted more rapidly are less abundant. This knowledge allows for further improvement of applications involving laccase such as the grafting of phenols on surfaces and enzymatic polymerization of lignin fragments.
Introduction
Laccases are copper-containing oxidoreductases found in plants, bacteria and fungi. In nature they act on catechols (1,2-dihydroxybenzenes) and other phenols and convert these to radical species via radical cation intermediates.1 Most members of the laccase family exhibit a very large substrate scope, which makes them useful catalysts in organic chemistry.2–4 This scope is enlarged even further when mediator systems are employed.5 In these cases a mediator molecule is first oxidized by laccase, after which this oxidizes a molecule that cannot directly be oxidized by laccase. This makes laccases interesting biocatalysts and underlines clearly why they are used in a wide variety of applications ranging from polymerization6–8 and small molecule synthesis9–12 to waste water purification13–15 and the modification of biorenewables.16,17 Within the sub-class of small molecule synthesis the formation of dimers out of (naturally occurring) phenols draws special attention.18–20For some of these transformations the molecular structure of the product(s) is discussed, mainly when it concerns single product reactions or polymerization reactions.21,22 However in many cases a particular transformation is only characterized by a qualitative description of observed changes in the system; for example a shift in the UV spectrum or disappearance of the starting material according to mass spectrometry. This is common when dye or drug degradation is studied.23,24
Apart from the previously mentioned wide variety of applications of laccase it is also known for its ability to graft small monomers from (natural) fibres such as wood or cotton.25–27 Within our group we developed a methodology to widen this scope even further by modifying poly(ethersulfone) (PES) membranes.28–32 In order to alter the surface of the membrane 4-hydroxybenzoic acid was grafted from the membrane via a laccase-mediated reaction. Through this environmentally benign process 4-hydroxybenzoic acid and a range of analogues thereof could be covalently attached to the otherwise rather inert membrane. This method can be used to change the surface properties of such membranes in a tailor-made fashion, e.g. making it anti-fouling.31 However, for further optimization of the anti-fouling properties a study of the molecular details of the grafting process is essential.
Although some studies have looked at the reaction products resulting from the laccase-mediated conversion of vanillic acid (4-hydroxy-3-methoxybenzoic acid) and oligomerization reactions of syringic acid (4-hydroxy-3,5-dimethoxybenzoic acid), further studies into the product profile for laccase-mediated reactions involving 4-hydroxybenzoic acid have not yet been performed.22,33 It could be that, in the case of vanillic and syringic acid, the additional methoxy substituents block positions on the benzene ring that would otherwise be available for C–C and C–O bond formation, which thereby makes the product profile for these substrates less complex and easier to study. Considering its importance as monomer in membrane modification reactions we aimed to study the laccase-mediated oligomerization of 4-hydroxybenzoic acid (a lignin fragment) in solution and provide detailed insight in the product profile of this reaction. In order to do so the reaction products were separated by analytical or preparative HPLC and subsequently analysed by mass spectrometry. The two major products were also analysed by NMR and their structure was proven through independent synthesis. Studies regarding the reactivity of these products were conducted subsequently.
Results and discussion
Trametes versicolor laccase is able to oxidize phenolic substrates to their radical cations, which rapidly lose a proton to form radical species that further react to oligomers under ambient conditions at pH 5. The reaction conditions were adopted from previous work related to the mentioned modification of PES membranes and employed in our current studies of the solution phase reaction of 4-hydroxybenzoic acid (4-HBA) and laccase.30 In the current study the buffer solution was changed from a sodium acetate/acetic acid buffer to an ammonium acetate/acetic acid buffer in order to be compatible with MS analysis. The reaction mixture of laccase and 4-HBA was mildly agitated for 24 h at room temperature with ambient air as oxidant. Thereafter laccase was removed from the mixture by centrifugal filtration through an ultrafiltration membrane to obtain enzyme-free material ready for LC-MS analysis.Determination of 4-hydroxybenzoic acid conversion
Earlier attempts to polymerize 4-hydroxybenzoic acid were reported to be unsuccessful,22 and it was claimed that 4-hydroxybenzoic acid was not oxidized under the used conditions at all (either of three different laccases in a mixed acetone/buffer medium). However, by employing, among others, colorimetric studies it was made clear that 4-hydroxybenzoic acid could be oxidized by laccase.30 Further research on neither the conversion nor the products from this reaction had been reported.In order to determine the amount of 4-hydroxybenzoic acid converted by the enzyme we measured the UV absorption of 4-hydroxybenzoic acid at 254 nm at several known concentrations. Using this calibration curve (ESI Fig. S1†) we determined the conversion by diluting an aliquot of the reaction mixture and again measured the absorption at 254 nm. From this we concluded that 26% 4-HBA was converted after 24 h using 26.8 mM 4-hydroxybenzoic acid and 1.0 U ml−1 of laccase.
Characterization of the main products of laccase-mediated oligomerization of 4-hydroxybenzoic acid
The follow-up goal was to determine the molecular structure of the main products of laccase-mediated oligomerization of 4-hydroxybenzoic acid. By employing LC-MS analysis we aimed to retrieve information on the molecular mass of the respective products and thereby an indication of their structure and a rough estimate of the relative abundance of the products. This information allows for a pre-selection of the most important molecules for further studies.Separation of the main products of laccase-mediated oligomerization of 4-hydroxybenzoic acid was achieved on a C18 column through a two-step isocratic elution with an acidified water/acetonitrile mixture. As was already anticipated, the reaction of 4-hydroxybenzoic acid with laccase resulted in a complex mixture of products (Fig. 1); more than 25 individual peaks are visible in the LC-UV chromatogram (254 nm).
 | ||
| Fig. 1 LC-UV chromatogram (254 nm) after the reaction of 4-hydroxybenzoic acid (peak at 3.7 min) and laccase. Insert: scale-out of the chromatogram for the section from 5.0 to 18.0 min. Reaction conditions: 4-hydroxybenzoic acid (18.5 mg, 0.13 mmol, 26.8 mM) and laccase (4.8 U, 1.0 U ml−1) in a 0.02 M ammonium acetate/acetic acid buffer (pH 5, 5.0 ml) mildly agitated at room temperature for 24 h. Separation was achieved through LC-method: LC1. | ||
A selection of the most important products was made based on the highest peaks in the chromatogram (Table 1). For all four components listed in Table 1 the corresponding molecular ion [M − H] was the signal with the highest intensity: adducts with other ions only made a minor contribution (see ESI Fig. S6–S12† for respective mass spectra and extracted ion chromatograms). Peak 1 at retention time 3.7 min corresponds to the starting material 4-hydroxybenzoic acid. The two product peaks with the highest UV absorption (peak 2 and 3 at retention time 4.5 min and 11.8 min), both correspond to a molecule with a mass of 274 Da. Furthermore, the third most intense product peak 4 has a retention time of 13.3 min and corresponds to a compound having a molecular mass of 380 Da. It is well known that laccase is able to oligomerize phenols and it is therefore logical that the products formed first are dimers that in our case would have a mass of 274 Da.21,33 These two expected dimers are most likely formed through either a carbon centered radical or an oxygen-centered radical, following a mechanism which is based on the proposed mechanism for other phenol dimerizations (Scheme 1).21,33 Laccase generates a 4-hydroxybenzoic acid radical cation that easily loses a proton. The thus formed radical is delocalized over the phenolic system. Two of these radicals can recombine following a tautomerization to form dimer 1 or 2 directly (top route). Alternatively, one of these radicals attacks a second molecule of 4-hydroxybenzoic acid to form another radical (propagation), from which a hydrogen atom is abstracted (termination) to form once again either dimer 1 or 2 (bottom route).
| # | RTa/min | [M − H]b/Da | Chem. for.c | Th. [M − H]d/Da |
|---|---|---|---|---|
| a Retention time. b Measured m/z with highest intensity ([M − H]) at respective retention time. c Putative chemical formula generated from measured [M − H]. d Theoretical [M − H] corresponding to putative chemical formula. | ||||
| 1 | 3.7 | 137.02311 | C7H5O3 | 137.02442 |
| 2 | 4.5 | 273.03978 | C14H9O6 | 273.04046 |
| 3 | 11.8 | 273.03971 | C14H9O6 | 273.04046 |
| 4 | 13.3 | 379.04394 | C20H11O8 | 379.04594 |
 | ||
| Scheme 1 Proposed pathway for the formation of dimers 1 and 2 through recombination of two 4-hydroxybenzoic acid radicals (top) or via radical propagation followed by proton abstraction (bottom). | ||
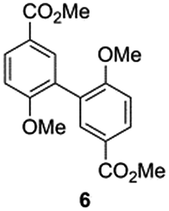
Independent synthesis of these dimers would facilitate their identification. The C3–C3′ coupled dimer 1 could be synthesized through an Ullmann coupling with two equivalents of methyl 3-iodo-4-methoxy-benzoate in the presence of copper bronze (Scheme 2).34 In order to obtain reasonable yields the reaction has to be heated above 250 °C and stirred vigorously in a shallow boiling tube. This process yielded 68% of methylated compound 6. Demethylation was thereafter achieved through the use of boron tribromide followed by continuous extraction of the very polar product dimer 1.35
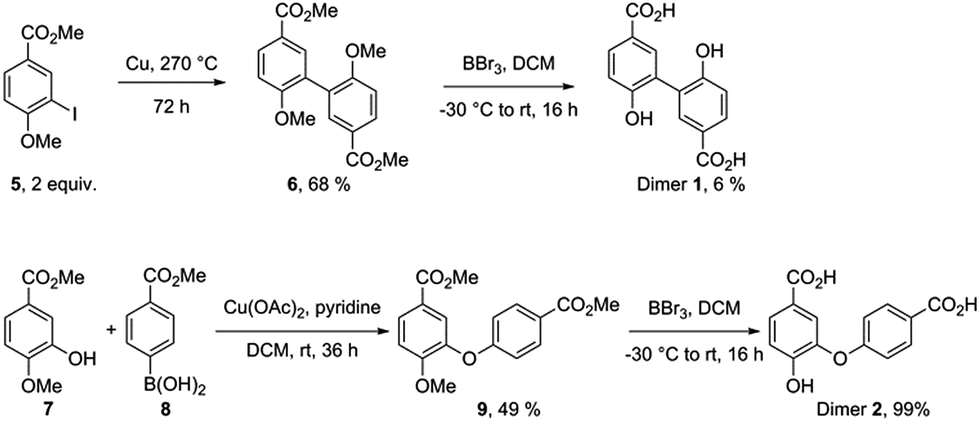 | ||
| Scheme 2 Synthesis route for dimer 1 (top) and dimer 2 (bottom). | ||
The C3–O coupled product 9 could be generated through a Chan-Lam coupling between commercially available methyl isovanillate and 4-methoxycarbonylphenylboronic acid in the presence of molecular sieves.36 Again, demethylation through the use of boron tribromide and continuous extraction resulted in the desired dimer.35 In order to properly compare retention times in HPLC at 24 h 4-hydroxybenzoic acid reaction sample was spiked with both synthesized dimers 1 and 2 and subsequently eluted under the same conditions as a non-spiked sample. The results (ESI Fig. S17†) make clear that the retention time for both dimers corresponds to the retention time of the putative dimers as reported in Table 1 (entry 2 and 3).
Furthermore, the mass spectra of the chemically synthesized dimers correspond to those of the dimers that are generated enzymatically (ESI Fig. S9, S10, S20 and S21†).
Definitive structure elucidation of the two putative dimers was achieved through their isolation from the reaction mixture by means of preparative HPLC. A comparison of 1H NMR data (ESI Table S2†) of both the chemically synthesized as well as the enzymatically generated material shows that the putative structures indeed correspond to the two main products in the laccase-mediated conversion of 4-hydroxybenzoic acid.
In order to obtain an even more complete characterization of the products originating from laccase oxidation of 4-hydroxybenzoic acid we also aimed to obtain information on the structure of minor products. The third most intense UV product peak corresponds to a compound with m/z = 379 (Table 1, entry 4). After analysing the corresponding putative chemical formula we concluded that this is most probably a trimeric benzoquinone (a non-exhaustive list of possible trimeric benzoquinone species is depicted in Scheme 3) in which one of the subunits is decarboxylated, eventually leading to a 1,2- or 1,4-benzoquinone. Formation of a benzoquinone is further supported by the occurrence of a second peak in the mass spectrum of this product at m/z = 381 (ESI Fig. S11 and S12†). This additional peak arises because the benzoquinone moiety is easily reduced, which is apparent in the formation of a diol moiety two mass units heavier than its corresponding benzoquinone. This reduction can take place at the spray tip of the electrospray ionization source of the mass spectrometer in negative ionization mode. Similar reduction processes have been observed for 1,2-benzoquinone and have been extensively reviewed.37,38
 | ||
| Scheme 3 Possible structures for a benzoquinone trimer. | ||
The formation of a benzoquinone is in line with previous findings involving vanillic acid as substrate, for which several benzoquinone-containing products were isolated.21,33 However, mechanistic details on this process remain until this point unclear. Where there are only limited options to couple two monomers, there is a multitude of options to couple three.
MS–MS analysis did not provide enough information to suggest a useful lead compound and more structural NMR-based information was required.
To obtain the material in sufficient quantities to be analysed by NMR the laccase-mediated modification of 4-hydroxybenzoic acid was performed at a 250 ml scale. Separation and purification of the individual components, with as main goal the isolation of one or more trimeric benzoquinones, would then be achieved through repetitive preparative HPLC-runs. However, 40 repetitive separations still did not provide enough material for proper NMR characterization. The trimeric benzoquinones appear to be only very minor constituents of the complex product mixture.
As we were unsuccessful with regards to isolating the suspected trimeric benzoquinone we did not try to isolate other minor products, since we expect those to be even less abundant.
Determination of conversion towards dimers 1 and 2
In a fashion similar to the determination of the conversion of 4-hydroxybenzoic acid, we were able to determine the yield of dimers 1 and 2 after laccase-mediated oligomerization of 4-HBA. After obtaining enough of both dimers through chemical synthesis as described above, we plotted a calibration curve (ESI Fig. S2 and S3†) for the UV absorption of the corresponding peaks after LC-separation at several known concentrations. By interpolation of the matching peak area of a 24 h laccase reaction sample to the calibration curve we determined that dimer 1 was obtained in 2% yield and dimer 2 in 0.2% yield. This means that the majority (almost 24%) of the previously mentioned 26% conversion of 4-HBA over 24 h is converted to other products. Most likely this is converted to a fast array of unique higher order oligomeric products which are all present in minute amounts. This composition therefore actually supports the efficient polymerization of 4-HBA and its success in membrane modification.Laccase-mediated transformation of dimers 1 and 2
The low abundance of the two dimers in the reaction mixture prompted us to further investigate the role of these products in the reaction pathway. We hypothesized that these were no final products but merely intermediates in a reaction mixture that is in a steady state in which the dimers are rapidly converted to different products, and in fact significantly faster than the monomer 4-hydroxybenzoic acid.To test this hypothesis we determined the conversion of both dimers under conditions similar to those of the reaction of laccase and 4-hydroxybenzoic acid. In this case the solubility of the dimers turned out to be the limiting factor, as a result we lowered the concentration of the starting dimer to 3.40 mM and changed the reaction medium to a 10% solution of methanol in 0.02 M ammonium acetate/acetic acid buffer (pH 5).
The conversion of dimers was determined under five different conditions in which either one of the two dimers was solely present as substrate in the reaction mixture, or together with the second dimer, or in a mixture with also 4-hydroxybenzoic acid present (Table 2).
| # | Substrates | Conv. dimer 1 | Conv. dimer 2 |
|---|---|---|---|
a Dimer(s) (1.4 mg, 5.0 μmol, 3.40 mM) and 4-hydroxybenzoic acid (4-HBA) (entry 4 and 5 only, 0.7 mg, 5.0 μmol, 3.40 mM) were reacted in the presence of laccase (0.51 U ml−1) in a mixture of methanol and a 0.02 M ammonium acetate/acetic acid buffer (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) at room temperature and pH 5 for 24 h. :9) at room temperature and pH 5 for 24 h.
|
|||
| 1 | Dimer 1 | 48% | — |
| 2 | Dimer 2 | — | 77% |
| 3 | Dimer 1 + dimer 2 | 53% | 69% |
| 4 | Dimer 1 + 4-HBA | 55% | — |
| 5 | Dimer 2 + 4-HBA | — | 69% |
As shown in Table 2, conversion of both dimers is similar across all screened conditions. Around 50% of dimer 1 and around 70–75% of dimer 2 is converted under these conditions. This is significantly higher than the conversion of 4-hydroxybenzoic acid after 24 h even under such non-optimal reaction conditions (lower concentration starting material and methanol as co-solvent). This supports the hypothesis that these dimers are highly reactive intermediate products in the laccase conversion pathway of 4-hydroxybenzoic acid. Furthermore the higher conversion of dimer 2 might also, in part, explain the lower yield of this dimer in laccase-mediated 4-hydroxybenzoic acid oligomerization as it is more rapidly converted than dimer 1.
In order to validate the hypothesis that dimers 1 and 2 are more reactive than the monomer 4-HBA we performed DFT calculations on the conversion of these substrates to their radical cation (ESI Table S1†). As depicted in Scheme 1 this is the rate-limiting step and will thereby most likely govern the reactivity of the substrates.
We found that the generation of the radical cation in water for 4-hydroxybenzoic acid occurs less easily than for both dimers (energy gap of 154.9 kcal mol−1 (4-HBA) vs. 152.7 kcal mol−1 (dimer 1) and 147.8 kcal mol−1 (dimer 2)). These results support our hypothesis that dimers 1 and 2 are highly reactive intermediates in the laccase-mediated conversion of 4-hydroxybenzoic acid.
Apart from some expected dimerization in the laccase-mediated treatment of dimer 2 towards a molecule with a molecular mass of 546 Da (ESI Fig. S13 and S14†) little information was obtained on the nature of the formed products from dimer conversion. In the condition where dimer 1 and dimer 2 were both present as a substrate, two more peaks (with very low intensity) corresponding to a molecule with molecular mass 546 Da were observed (data not shown). Again (as with the conversion of 4-HBA) a wide range of molecules seem to be formed albeit all in very low yields.
The role of less abundant products
As indicated by the rapid conversion of dimers 1 and 2, products with low abundance might still be important in the oligomerization process. This implies that the pre-selection based on peak intensity will not include all relevant compounds. To find out what other molecules might play a role, a close look was taken into the mechanism of dimer formation (Scheme 1).So far the contributions of radicals A˙ and B˙ to form dimers 1 and 2 have been taken into account. Radical C˙ however might also give rise to dimers followed by decarboxylation. In this case dimers 3 and 4 are formed by attack of radical C˙ on a molecule of 4-HBA, another radical A˙ or radical B˙ (Scheme 4). Similar as to the identification of dimers 1 and 2 we now aimed to obtain and detect possible dimers 3 and 4 to find out if these also play a role in the effective oligomerization of 4-HBA by laccase.
 | ||
| Scheme 4 Dimers possibly originating from radical C˙ (230 Da). | ||
These dimers will most likely have a stronger affinity for the used C18 column during LC-separation, therefore the LC-method was slightly adjusted towards a higher percentage of the less polar solvent acetonitrile (LC3). Indeed several peaks that correspond to a molecule having a mass of 230 Da were observed (ESI Fig. S15†). Some of these peaks represent MS-generated adducts or fragments of molecules which are present in far higher concentrations, i.e. the peak at 10.6 min which represents a fragment of dimer 2. The peak at 7.7 min (mass spectrum in ESI Fig. S16†), however, might correspond to one of the putative dimers.
In order to test this hypothesis we followed a similar approach as to the identification of dimers 1 and 2. For this purpose, chemically synthesized dimers need to be obtained in order to compare their retention times with those corresponding to the putative dimeric structures. Due to the even lower apparent abundance (based on UV intensity) of the putative dimers 3 and 4 compared to that of the possible benzoquinone (i.e. compound 10, 11 or 12), isolation through preparative HPLC was not pursued.
Whereas dimer 4 is commercially available, dimer 3 was synthesized using a slight modification of a recently published one-pot two-step synthesis involving a Suzuki coupling followed by saponification (Scheme 5).39
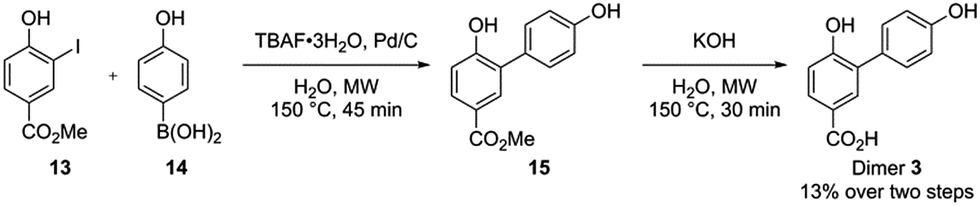 | ||
| Scheme 5 Synthesis route for dimer 3. | ||
Co-injection of the dimers with an aliquot from the reaction of 4-HBA with laccase indicated the presence of dimer 3 (ESI Fig. S18†). Dimer 4, however, was difficult to observe (ESI Fig. S19†). Over the course of the reaction the yield of dimer 3 fluctuated between approximately 0.01% and 0.02% (ESI Fig. S23†). The fact that these yields are so low indicates a rapid oligomerization of dimers 3 and 4.
To test this hypothesis, as was done for dimers 1 and 2 before, the laccase-mediated conversion of dimers 3 and 4 was monitored after 24 h. Yet again the solubility, especially of dimer 4, proved to be an issue. This was overcome by increasing the percentage of the co-solvent methanol in the reaction mixture to 18.5%. As shown in Table 3 the conversion of dimer 3 went to completion over the course of 24 h (entry 1 and 3). This was also true for dimer 4 when it was present as sole substrate (entry 2). However, when 4-hydroxybenzoic acid was also present, only 70% of dimer 4 was converted (entry 4).
| # | Substrates | Conv. dimer 3 | Conv. dimer 4b |
|---|---|---|---|
|
a Dimer 3 or 4 (3.40 mM) and 4-hydroxybenzoic acid (4-HBA) (entry 3 and 4 only, 3.40 mM) were reacted in the presence of laccase (0.51 U ml−1) in a mixture of methanol and a 0.02 M ammonium acetate/acetic acid buffer (1:4.4) at room temperature and pH 5 for 24 h.
b The determined conversions of dimer 4 are an average of 3 measurements.
|
|||
| 1 | Dimer 3 | >99% | — |
| 2 | Dimer 4 | — | 99% |
| 3 | Dimer 3 + 4-HBA | >99% | — |
| 4 | Dimer 4 + 4-HBA | — | 70% |
Full conversion of dimer 3 through laccase-mediated oligomerization explains the finding that the yield of this dimer resulting from the laccase-mediated oligomerization of 4-HBA is so low. This is, most likely, also true for dimer 4 when one takes into account the even less optimal conditions (18.5% MeOH) under which these reactions have been performed.
Based purely on the conversion at 24 h it is not possible to differentiate between the rates for the reaction of laccase with dimer 3 or 4. The conversion of both dimers, with or without 4-HBA, was therefore also monitored over time. The conversion of dimer 3 occurred with similar rates in both presence and absence of 4-HBA, and is already almost complete at 4 h (ESI Fig. S24†). In the absence of 4-HBA, dimer 4 seems to be converted slightly slower than dimer 3 (ESI Fig. S25†). When 4-hydroxybenzoic acid is present, however, the reaction does not seem to obey regular kinetics. The reason for this is unclear. Considering the structural similarity between both, it could be that 4-HBA acts as a competitive inhibitor for the conversion of dimer 4.
The relative reactivities of each dimer were also estimated by calculating the energy required for the formation of the radical cation from the respective dimer (ESI Table S1†). These results (dimer 3: 137.1 kcal mol−1; dimer 4: 138.1 kcal mol−1) clearly underline that dimers 3 and 4 are even more reactive than dimers 1 and 2 and thus than 4-HBA. Apart from this, the subtle differences in calculated relative energies are reflected in the rate of reaction of each dimer (dimer 3 > dimer 4 ≫ dimer 2 ≫ dimer 1 ≫ 4-HBA).
These results clearly indicate that the yield of dimers is largely governed by rapid subsequent oligomerization. Furthermore, both the theoretical calculations and the experimental evidence thus support our hypothesis that the formed dimers are not final products, but merely intermediates in the laccase-mediated conversion of 4-hydroxybenzoic acid.
Conclusions
Laccase-mediated oligomerization of 4-hydroxybenzoic acid results in a wide variety of products. The moderately reactive monomer 4-HBA is first converted to at least three distinct dimers in which the monomers are bound via either a C3–C3′, C3–O or C1–C3′ linkage. Both theoretical as well as experimental evidence showed that these dimers were converted far more rapidly than 4-HBA itself. This is reflected in the yield of the above mentioned dimers: the more reactive, the lower the yield. These results indicate that the dimers are not final products of the reaction of 4-hydroxybenzoic acid with laccase, but highly reactive intermediates in the efficient formation of polymers.The potential for covalent modification of poly(ethersulfone) membranes by such polymers clearly highlights the importance of these findings. We expect that the newly obtained knowledge will open up avenues towards discovering new, and improving current, applications in the laccase-mediated modification of phenols and other lignin fragments.
Experimental
Equipment and general conditions
Chemicals: 4-hydroxybenzoic acid (>99%), methyl 4-hydroxy-3-iodobenzoate (97%), palladium on carbon (10 wt%), potassium hydroxide (≥85%), Trametes versicolor (EC number: 1.10.3.2) were obtained from Sigma-Aldrich; methyl 3-iodo-4-methoxybenzoate (98%), copper acetate (98%), copper bronze powder (99%) came from Alfa Aesar; methyl 3-hydroxy-4-methoxybenzoate (>98%), 4-(4-hydroxyphenoxy)benzoic acid (>99%) were obtained from TCI Chemicals, 4-methoxycarbonylphenylboronic acid (97%) was purchased from Fluorochem, ABTS diammonium salt (98%) was obtained from Amresco, boron tribromide (>99%), 4-hydroxyphenylboronic acid (97%) were obtained from Acros Organics, tetrabutylammonium fluoride trihydrate (for synthesis) was purchased from Merck Millipore, glacial acetic acid (ULC/MS) was obtained from Biosolve and ammonium acetate (analytical grade) was obtained from Fisher Scientific. Reactions were performed under ambient conditions unless stated otherwise. Reactions heated by microwave irradiation were performed in a Anton-Paar Monowave 400 microwave with internal temperature control. Purification by flash chromatography was conducted by using flash silica gel 60. 1H and 13C spectra were recorded on a 400 MHz Bruker Avance spectrometer. All signals were referenced relative to the residual solvent signal and coupling constants J are given in Hz. High resolution mass spectra were recorded on a Thermo Scientific Exactive instrument. All MS chromatograms were normalized to the peak with highest intensity. Extracted ion chromatograms are always displayed as a plot of [M − H] ± 0.50 Da.Enzyme assay
The specific activity of commercially available laccase from Trametes versicolor (EC number: 1.10.3.2) was determined spectroscopically on a Varian Cary 50 Scan UV/Vis spectrometer by measuring the decrease of absorbance for the oxidation of ABTS to the ABTS radical cation (ε = 36000). ABTS diammonium salt was dissolved in 0.02 M ammonium acetate/acetic acid buffer (pH 5, 3.5 ml) and laccase was added after which the decrease of absorbance at λ = 420 nm was followed for 1.0 min. The specific activity was determined to be 3.2 U mg−1 laccase.40
Laccase-mediated conversion of 4-hydroxybenzoic acid
4-Hydroxybenzoic acid (18.5 mg, 0.13 mmol, 26.8 mM) and laccase from Trametes versicolor (EC number: 1.10.3.2, 4.8 U, 1.0 U ml−1) were dissolved in 0.02 M ammonium acetate/acetic acid buffer (pH 5, 5.0 ml) and mildly agitated at room temperature for 24 h. Thereafter laccase was removed from the reaction mixture using a 9 kDa protein concentrator by employing centrifugal force (Hermle Z 206A). The supernatant was then either diluted in buffer and subjected to LC-MS analysis or its constituents were separated by employing preparative HPLC.Laccase-mediated conversion of dimers
Dimers (3.40 mM each) were dissolved in MeOH (10% for dimers 1 and 2 or 18.5% for dimers 3 and 4) and mixed with either 0.02 M ammonium acetate/acetic acid buffer (pH 5) or a solution of 4-hydroxybenzoic acid (3.40 mM) in 0.02 M ammonium acetate/acetic acid buffer (pH 5). To this a solution of laccase from Trametes versicolor (EC number: 1.10.3.2) in buffer was added (0.51 U ml−1) after which the reaction mixture was mildly agitated at room temperature. Subsequent removal of laccase employing centrifugal force (10 protein concentrator, Hermle Z 206A) was followed by appropriately diluting the supernatant with buffer, after which the mixture was subjected to LC-MS analysis.Analytical LC-MS setup
For liquid chromatography/mass spectrometry (LC-MS) a Thermo Finnigan Surveyor MS pump was coupled to a photodiode array detector (Thermo Finnigan Surveyor PDA) and either one of the two following: a mass spectrometer (Finnigan LXQ) or high resolution mass spectrometer (Thermo Scientific Exactive), and both instruments were equipped with an electrospray ionization source. Measurements were conducted in negative ionization mode. LC-MS-MS measurements were conducted with the Finnigan LXQ mass spectrometer where components were fragmented at a normalized collision energy of 35 eV. Separation was achieved through an Alltima HP C18 column (2.1 mm × 100 mm, 3 μm; Grace) at room temperature and a flow rate of 0.2 ml min−1. The mobile phases consisted of 0.1% formic acid in water (mobile phase A) and in acetonitrile (mobile phase B).The used method for separation of the products of 4-hydroxybenzoic acid oligomerization was as follows: isocratic elution for 5.0 min at 15% B followed by isocratic elution for 12.0 min at 20% B after which a steep gradient towards 100% B within 0.1 min was followed by isocratic elution for 13.0 min at 100% B. This was followed by a steep gradient back to 15% B and the system was then re-equilibrated by employing 15.0 min of 15% B (LC1).
The used method for separation of the products of the laccase-mediated transformation of dimers 1 and 2 was as follows: gradient elution starting at 15% B which increases linearly to 50% B over the course of 20.0 min. This was followed by eluting with 100% B for 10.0 min after which the system was re-equilibrated by employing 15% B for 15.0 min (LC2).
The used method for detection of decarboxylated products from the laccase-mediated conversion of 4-HBA was as follows: isocratic elution for 5.0 min at 15% B directly followed by a linear increase of 20% B to 40% B over the course of 12.0 min. This was followed by eluting with 100% B for 6 or 15.0 min (dependent on the dilution of the injected sample) after which the system was re-equilibrated by employing 15% B for 13.0 min (LC3).
Preparative HPLC-setup
Preparative HPLC-separations were performed on an Alltima C18 column (22 mm × 250 mm, 5 μm; Grace). A LC-8A Shimadzu LC-pump was connected to a SPD-M10AVP Shimadzu diode array detector equipped with a FRC-10A Shimadzu fraction collector. Separation was achieved at room temperature at a constant flow of 22 ml min−1 using a mixture of 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in acetonitrile (mobile phase B). The used method was as follows: isocratic elution for 17.0 min at 22.5% B followed by an 11.0 min rinsing step of 100% B to finish with re-equilibration at 22.5% B.Quantitative NMR analysis
The purity of the synthesized dimers was assessed using quantitative NMR analysis. Either of the dimers and the internal standard 3,5-dinitrobenzoic acid were dissolved in 0.6 ml deuterated methanol (for dimer 1) or deuterated acetone (for dimers 2 and 3). Thereafter 1H-NMR spectra were recorded with high relaxation delay d1 (10 seconds). The purity of the synthesized dimer could then be determined according to the formula % purity by weight = Wis/Ws × As/Ais × MWs/MWis × His/Hs. In this formula W stands for the weighed amount of internal standard/dimer; A corresponds to the area of the particular NMR peak, MW stands for the molecular weight of the internal standard/dimer and H stands for the number of protons represented by the integral of the NMR peak.Determination of conversion
The peak area for the signal corresponding to the molecule of interest (UV, 254 nm) was determined after eluting samples with a known concentration of the specific target compound at the conditions as stated for the analytical LC-MS setup. This peak area was determined at several known concentrations (see ESI Fig. S1–S5† for details). The peak area at 0.00 μM was subtracted from the other data points and a line was fitted through the points using the least squares method (calibration curve). The obtained values for the slope (a) and offset (b) were used in the formula with form y = ax + b where x is the concentration of the specific target compound and y is the measured peak area.To determine the unknown concentration of the compound of interest after reaction an aliquot of the reaction mixture was diluted appropriately in order for the concentration to be interpolated on the calibration curve. This diluted aliquot was eluted under the same conditions as described above and the peak area for the UV peak (254 nm) corresponding to the target compound was determined. This obtained y was then used to regressively calculate the concentration of the compound of interest and thereby also the conversion of starting material.
Synthesis
 :60/ethyl acetate 6:1) to yield the product as an off-white solid (192 mg, 0.57 mmol, 68%). 1H NMR (400 MHz, chloroform-d): δ = 8.06 (dd, 3J(H,H) = 8.7, 2.3 Hz, 2H), 7.92 (d, 3J(H,H) = 2.2 Hz, 2H), 6.99 (d, 3J(H,H) = 8.7 Hz, 2H), 3.88 (s, 6H), 3.82 (s, 6H). 13C NMR (101 MHz, chloroform-d): δ = 167.0, 161.0, 133.1, 131.4, 126.9, 122.4, 110.5, 56.0, 52.1; HRMS (ESI) m/z [M + Na]+ 353.0992 (calcd. 353.0996).
:60/ethyl acetate 6:1) to yield the product as an off-white solid (192 mg, 0.57 mmol, 68%). 1H NMR (400 MHz, chloroform-d): δ = 8.06 (dd, 3J(H,H) = 8.7, 2.3 Hz, 2H), 7.92 (d, 3J(H,H) = 2.2 Hz, 2H), 6.99 (d, 3J(H,H) = 8.7 Hz, 2H), 3.88 (s, 6H), 3.82 (s, 6H). 13C NMR (101 MHz, chloroform-d): δ = 167.0, 161.0, 133.1, 131.4, 126.9, 122.4, 110.5, 56.0, 52.1; HRMS (ESI) m/z [M + Na]+ 353.0992 (calcd. 353.0996). :60/ethyl acetate 4:1) to yield the product as a white solid (170 mg, 0.54 mmol, 49%). 1H NMR (400 MHz, chloroform-d): δ = 8.01–7.94 (m, 2H), 7.92 (dd, 3J(H,H) = 8.6, 2.1 Hz, 1H), 7.73 (d, 3J(H,H) = 2.1 Hz, 1H), 7.03 (d, 3J(H,H) = 8.7 Hz, 1H), 6.94–6.84 (m, 2H), 3.87 (s, 3H), 3.85 (s, 3H), 3.83 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ = 166.6, 166.1, 161.7, 155.5, 143.0, 131.6, 128.1, 124.3, 123.4, 123.3, 115.9, 112.05, 56.0, 52.1, 52.0; HRMS (ESI) m/z [M + Na]+ 339.0831 (calcd. 339.0839).
:60/ethyl acetate 4:1) to yield the product as a white solid (170 mg, 0.54 mmol, 49%). 1H NMR (400 MHz, chloroform-d): δ = 8.01–7.94 (m, 2H), 7.92 (dd, 3J(H,H) = 8.6, 2.1 Hz, 1H), 7.73 (d, 3J(H,H) = 2.1 Hz, 1H), 7.03 (d, 3J(H,H) = 8.7 Hz, 1H), 6.94–6.84 (m, 2H), 3.87 (s, 3H), 3.85 (s, 3H), 3.83 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ = 166.6, 166.1, 161.7, 155.5, 143.0, 131.6, 128.1, 124.3, 123.4, 123.3, 115.9, 112.05, 56.0, 52.1, 52.0; HRMS (ESI) m/z [M + Na]+ 339.0831 (calcd. 339.0839).
General procedure for demethylation
A solution of the methylated precursor (0.6 mmol, 1 eq.) in dichloromethane (5 ml) was loaded in a three-necked flask on which an acid gas trap was fixed.35 The solution was stirred under N2 and cooled to −30 °C after which BBr3 (1.2 ml, 12.8 mmol, 20 eq.) was slowly added. Thereafter the mixture was further stirred for 18 h during which the mixture was allowed to warm up to room temperature. The reaction was quenched by carefully adding cold water (1 ml). The mixture was concentrated in vacuo after which it was continuously extracted using ethyl acetate (80 ml). The obtained solid was further purified using either silica gel chromatography or reversed phase preparative HPLC.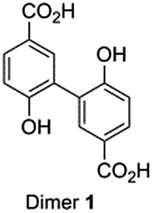
 :60/ethyl acetate/glacial acetic acid 1:5:0.02) to yield dimer 2 as an off-white solid (179 mg, 0.60 mmol*, 99%*, purity: 93% (q-NMR)). 1H NMR (400 MHz, acetone-d6): δ = 8.03 (d, 3J(H,H) = 8.8 Hz, 2H), 7.85 (dd, 3J(H,H) = 8.5, 2.0 Hz, 1H), 7.70 (d, 3J(H,H) = 2.0 Hz, 1H), 7.16 (d, 3J(H,H) = 8.5 Hz, 1H), 7.02 (d, 3J(H,H) = 8.8 Hz, 2H). 13C NMR (101 MHz, acetone-d6): δ = 167.1, 166.8, 162.6, 154.6, 142.7, 132.6, 129.1, 125.8, 124.4, 123.9, 118.0, 117.0; HRMS (ESI) m/z [M − H]− 273.0405 (calcd. 273.0405). * Based on 93% purity material.
:60/ethyl acetate/glacial acetic acid 1:5:0.02) to yield dimer 2 as an off-white solid (179 mg, 0.60 mmol*, 99%*, purity: 93% (q-NMR)). 1H NMR (400 MHz, acetone-d6): δ = 8.03 (d, 3J(H,H) = 8.8 Hz, 2H), 7.85 (dd, 3J(H,H) = 8.5, 2.0 Hz, 1H), 7.70 (d, 3J(H,H) = 2.0 Hz, 1H), 7.16 (d, 3J(H,H) = 8.5 Hz, 1H), 7.02 (d, 3J(H,H) = 8.8 Hz, 2H). 13C NMR (101 MHz, acetone-d6): δ = 167.1, 166.8, 162.6, 154.6, 142.7, 132.6, 129.1, 125.8, 124.4, 123.9, 118.0, 117.0; HRMS (ESI) m/z [M − H]− 273.0405 (calcd. 273.0405). * Based on 93% purity material.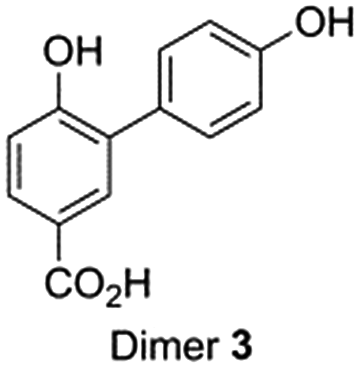 :2:0.1% to 1:1:0.1%) afforded the product as blue/white solid (9 mg, 0.04 mmol*, 13%*, purity: 96% (q-NMR)). 1H NMR (400 MHz, acetone-d6): δ = 9.02 (br s, 1H), 7.97 (d, J = 2.2 Hz, 1H), 7.84 (dd, J = 8.4, 2.2 Hz, 1H), 7.46 (dt, J = 8.8, 2.9, 2.1 Hz, 2H), 7.04 (d, J = 8.4 Hz, 1H), 6.91 (dt, J = 8.6, 2.9, 2.2 Hz, 2H). 13C NMR (101 MHz, acetone-d6): δ = 167.5, 159.2, 157.7, 133.2, 131.3, 130.8, 129.8, 129.3, 123.2, 116.7, 115.9. HRMS (ESI) m/z [M − H]− 229.0498 (calcd. 229.0506). * Based on 96% purity material.
:2:0.1% to 1:1:0.1%) afforded the product as blue/white solid (9 mg, 0.04 mmol*, 13%*, purity: 96% (q-NMR)). 1H NMR (400 MHz, acetone-d6): δ = 9.02 (br s, 1H), 7.97 (d, J = 2.2 Hz, 1H), 7.84 (dd, J = 8.4, 2.2 Hz, 1H), 7.46 (dt, J = 8.8, 2.9, 2.1 Hz, 2H), 7.04 (d, J = 8.4 Hz, 1H), 6.91 (dt, J = 8.6, 2.9, 2.2 Hz, 2H). 13C NMR (101 MHz, acetone-d6): δ = 167.5, 159.2, 157.7, 133.2, 131.3, 130.8, 129.8, 129.3, 123.2, 116.7, 115.9. HRMS (ESI) m/z [M − H]− 229.0498 (calcd. 229.0506). * Based on 96% purity material.
Quantum chemical calculations
Before performing theoretical calculations an initial conformational search was executed in order to determine the conformer distribution for both dimers and 4-HBA using Spartan'14. Hereby a constraint free search was performed and the resulting obtained structures were optimized using the MMFF force field. For all structures the conformer with the lowest energy was chosen for further studies at M11L level. These geometries were optimized using density functional theory (M11L with the 6-311+G(d,p) basis set) in water as solvent using Gaussian 09, this geometrical optimization was also applied to the radical cations.41 Additionally, optimization of all geometries was also performed at the B3LYP and M11L level in vacuum with similar results albeit with higher relative energies.Acknowledgements
This project was funded by the NanoNextNL project 04A.01 “Biomimetic selective layers”.References
- V. Madhavi and S. S. Lele, BioResources, 2009, 4, 1694–1717 Search PubMed.
- M. Mogharabi and M. A. Faramarzi, Adv. Synth. Catal., 2014, 356, 897–927 CrossRef CAS.
- S. Riva, Trends Biotechnol., 2006, 24, 219–226 CrossRef CAS PubMed.
- S. Witayakran and A. J. Ragauskas, Adv. Synth. Catal., 2009, 351, 1187–1209 CrossRef CAS.
- R. Reiss, J. Ihssen, M. Richter, E. Eichhorn, B. Schilling and L. Thöny-Meyer, PLoS One, 2013, 8, e65633 CAS.
- F. Hollmann and I. W. C. E. Arends, Polymers, 2012, 4, 759–793 CrossRef.
- S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353 CrossRef CAS PubMed.
- Y. Zhang, X. Fan, Q. Wang and A. Cavaco-Paulo, RSC Adv., 2016, 6, 49272–49280 RSC.
- G. A. Kraus, A. Thite and F. Liu, Tetrahedron Lett., 2009, 50, 5303–5304 CrossRef CAS.
- S. Hajdok, J. Conrad, H. Leutbecher, S. Strobel, T. Schleid and U. Beifuss, J. Org. Chem., 2009, 74, 7230–7237 CrossRef CAS PubMed.
- J. Gonzalez-Sabin, N. Rios-Lombardia, I. Garcia, N. M. Vior, A. F. Brana, C. Mendez, J. A. Salas and F. Moris, Green Chem., 2016, 18, 989–994 RSC.
- E. Ricklefs, M. Girhard, K. Koschorreck, M. S. Smit and V. B. Urlacher, ChemCatChem, 2015, 7, 1857–1864 CrossRef CAS.
- P. Demarche, C. Junghanns, R. R. Nair and S. N. Agathos, Biotechnol. Adv., 2012, 30, 933–953 CrossRef CAS PubMed.
- S. Dong, H. Xiao, Q. Huang, J. Zhang, L. Mao and S. Gao, Sci. Rep., 2016, 6, 21396 CrossRef CAS PubMed.
- X. Zhang, M. Hua, L. Lv and B. Pan, Sci. Rep., 2015, 5, 8253 CrossRef CAS PubMed.
- M. C. R. Franssen, P. Steunenberg, E. L. Scott, H. Zuilhof and J. P. M. Sanders, Chem. Soc. Rev., 2013, 42, 6491–6533 RSC.
- A. Rico, J. Rencoret, J. C. del Río, A. T. Martínez and A. Gutiérrez, Biotechnol. Biofuels, 2014, 7, 1–14 CrossRef PubMed.
- S. Ncanana, L. Baratto, L. Roncaglia, S. Riva and S. G. Burton, Adv. Synth. Catal., 2007, 349, 1507–1513 CrossRef CAS.
- O. E. Adelakun, T. Kudanga, A. Parker, I. R. Green, M. le Roes-Hill and S. G. Burton, J. Mol. Catal. B: Enzym., 2012, 74, 29–35 CrossRef CAS.
- E. Beneventi, S. Conte, M. R. Cramarossa, S. Riva and L. Forti, Tetrahedron, 2015, 71, 3052–3058 CrossRef CAS.
- A. Leonowicz, R. U. Edgehill and J. M. Bollag, Arch. Microbiol., 1984, 137, 89–96 CrossRef CAS.
- R. Ikeda, J. Sugihara, H. Uyama and S. Kobayashi, Polym. Int., 1998, 47, 295–301 CrossRef CAS.
- S. Camarero, D. Ibarra, M. J. Martínez and Á. T. Martínez, Appl. Environ. Microbiol., 2005, 71, 1775–1784 CrossRef CAS PubMed.
- L. Lloret, G. Eibes, T. A. Lú-Chau, M. T. Moreira, G. Feijoo and J. M. Lema, Biochem. Eng. J., 2010, 51, 124–131 CrossRef CAS.
- J. Rencoret, E. Aracri, A. Gutiérrez, J. C. del Río, A. L. Torres, T. Vidal and A. T. Martínez, Biochem. Eng. J., 2014, 86, 16–23 CrossRef CAS.
- T. Kudanga, G. S. Nyanhongo, G. M. Guebitz and S. Burton, Enzyme Microb. Technol., 2011, 48, 195–208 CrossRef CAS PubMed.
- R. P. Chandra, C. Felby and A. J. Ragauskas, J. Wood Chem. Technol., 2005, 24, 69–81 CrossRef.
- S. van der Veen, N. Nady, M. C. R. Franssen, H. Zuilhof, R. M. Boom, T. Abee and K. Schroën, J. Appl. Polym. Sci., 2015, 132, 41576 CrossRef.
- N. Nady, M. C. R. Franssen, H. Zuilhof, R. M. Boom and K. Schroën, Water, 2012, 4, 932–943 CrossRef CAS.
- N. Nady, K. Schroën, M. C. R. Franssen, M. S. Mohyeldin, H. Zuilhof and R. M. Boom, J. Membr. Sci., 2012, 394, 69–79 CrossRef.
- N. Nady, K. Schroën, M. C. R. Franssen, R. Fokkink, M. S. Mohyeldin, H. Zuilhof and R. M. Boom, J. Colloid Interface Sci., 2012, 378, 191–200 CrossRef CAS PubMed.
- N. Nady, K. Schroën, M. C. R. Franssen, B. van Lagen, S. Murali, R. M. Boom, M. S. Mohyeldin and H. Zuilhof, ACS Appl. Mater. Interfaces, 2011, 3, 801–810 CAS.
- J.-M. Bollag, S.-Y. Liu and R. D. Minard, Soil Biol. Biochem., 1982, 14, 157–163 CrossRef CAS.
- D. Drochner, W. Hüttel, S. E. Bode, M. Müller, U. Karl, M. Nieger and W. Steglich, Eur. J. Org. Chem., 2007, 2007, 1749–1758 CrossRef.
- J. F. W. McOmie, M. L. Watts and D. E. West, Tetrahedron, 1968, 24, 2289–2292 CrossRef CAS.
- S. M. Johnson, S. Connelly, I. A. Wilson and J. W. Kelly, J. Med. Chem., 2008, 51, 6348–6358 CrossRef CAS PubMed.
- N. B. Cech and C. G. Enke, Mass Spectrom. Rev., 2001, 20, 362–387 CrossRef CAS PubMed.
- G. Albarran, W. Boggess, V. Rassolov and R. H. Schuler, J. Phys. Chem. A, 2010, 114, 7470–7478 CrossRef CAS PubMed.
- B. Schmidt and M. Riemer, Eur. J. Org. Chem., 2015, 2015, 3760–3766 CrossRef CAS.
- F. Arnold, G. Georgiou, M. Alcalde and T. Bulter, in Directed Enzyme Evolution, Humana Press, 2003, vol. 230, pp. 193–201 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra23040g |
| This journal is © The Royal Society of Chemistry 2016 |