
Effect of nitrogen substitution on the structural and magnetic ordering transitions of NiCr2O4†
Xin Liu*ab,
Nan Yina,
Tiju Thomasc,
Minghui Yang*a,
Junhu Wangab and
Quan Shi*a
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: shiquan@dicp.ac.cn; myang@dicp.ac.cn; liuxin01@dicp.ac.cn
bMössbauer Effect Data Center, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
cDepartment of Metallurgical and Materials Engineering, Indian Institute of Technology Madras, Chennai 600036, Tamil Nadu, India
First published on 21st November 2016
Abstract
The nitrogen (N) doped spinel NiCr2O4 has been synthesized at 773 K (N500) and 873 K (N600) by ammonolysis of NiCr2O4 powders to study the effect of anion doping on its structural and magnetic properties. The N contents are determined by thermogravimetric oxidation, yielding a composition that can be described as NiCr2O3.68N0.21 (N500) and NiCr2O3.55N0.30 (N600). X-ray photoelectron spectroscopic studies suggest that N3− species partly substitute the oxygen in the lattice and oxygen vacancies exist in the N doped samples. There is evidence that in the nitrided sample, the Cr ion is most likely in a mixed oxidation state. As the N content increases, the structure at room temperature changes from tetragonal to the cubic phase; N500 is only partially tetragonal; N600 is completely cubic. Such structural change is the consequence of the depression of the cooperative Jahn–Teller effect of Ni2+ in the tetrahedral A site caused by the presence of N3−. Combined heat capacity and temperature dependent magnetic susceptibility measurements give clear evidence of the magnetic and structural transitions in the N doped NiCr2O4. The Jahn–Teller transition temperatures decrease with increasing N content; this is likely due to increased covalency and hence enhanced contribution of the angular momentum and the spin–orbit coupling to local chemical bonding around Ni2+. Antiferromagnetic transitions are observed at TS = 23 K and 22 K for N500 and N600, respectively. Hence there is indeed a lowering of transition when compared to pure NiCr2O4 (28 K). The magnetic loops at different temperatures confirm that the material behaves as a paramagnet over a wide range of temperatures T ∼ 80–350 K. The material also exhibits a canted ferrimagnetic structural transition between 30 and 70 K. We also report evidence for increased frustration and lowered correlation length in N doped compounds compared to the parent NiCr2O4. The present study on N− doping effects on the structure and magnetic properties of this NiCr2O4 is expected to be useful for tailoring the ferric phase transitions through anion substitutions.
1 Introduction
Nickel chromite (NiCr2O4) is a normal spinel and a common mineral in Earth's mantle and crust.1,2 It is commonly used for catalytic applications3–6 and gas sensors.7 It is also a well known magnetodielectric material that was first reported by Mufti et al.8 The spinel NiCr2O4 exhibits interesting structural–magnetic correlation.9,10 In the cubic structure, Cr3+ (with 3d3 configuration) preferentially occupies the octahedral sites because of the strong crystal field stabilization of the half filled non-degenerate t2g orbitals and empty eg orbitals, while the Ni2+ (with 3d8 configuration) are found in the tetrahedral sites.11 The tetrahedral crystal field around Ni2+ (3d8) in the cubic phase results in fully occupied low energy e levels and triply degenerate t2 levels, rendering this structure potentially unstable. Consequently, a cooperative lattice distortion – from cubic to tetragonal symmetry – lifts the orbital degeneracy in NiCr2O4 at 310 K.12 This tetrahedral distortion results in the elongation of NiO4 tetrahedra and c/a > 1. NiCr2O4 shows the occurrence of two magnetic transitions – TC and TS, corresponding to the onset of a ferrimagnetic (longitudinal) component and an antiferromagnetic (transverse) component.13 By using neutron scattering, Ishibashi and Yasumi have confirmed a further structural distortion from tetragonal symmetry to orthorhombic symmetry at 65 K, which is correlated with the onset of ferrimagnetic ordering.14,15 At lower temperatures, the NiCr2O4 exhibits a canted magnetic structure in which the ferromagnetic component and the antiferromagnetic (AF) component forms is consistent with long-range order below TS = 31 K. A distortion within the orthorhombic structure takes place at the same temperature, where anomalies in magnetic susceptibility and heat capacity are found.16,17 The investigation of temperature and magnetic field dependence of dielectric properties reveals a new anomaly at 20 K, corresponding to the completion of structural and magnetic order.10Considering the rich structural chemistry of NiCr2O4 a lot of research efforts are ongoing to do with tailoring the phase transitions. Most of the work has been focused on modifications of cation composition, through doping of Cu,18,19 Co,20–22 Mg,23 Fe,24 etc. into the NiCr2O4 lattice. In effect these studies explore the effect of the transition metal cations with different numbers of d electrons on observed structural distortions and the magnetic transition temperatures. A less explored approach is to investigate impact of anion composition on the structural transitions in NiCr2O4. Morris et al. reported that NiCr2S4 and NiCr2Se4 both have the Cr3S4 structure.25 It is relevant to note that the Cr3S4 (I2/m) system does not have a spinel structure, and that is not antiferromagnetically ordered at low temperatures. In fact in this system, the order moments are reduced from spin only values, due to the appreciable covalency of the metal–sulfur bonds.26 Hence the impacts of anion doping in an oxide material such as NiCr2O4 is certainly interesting.
Transition-metal oxides can be transformed into oxynitrides by reacting with ammonia at elevated temperatures. From the structural point of view, oxynitrides often show the same structural type as the parent oxides for moderate substitutional rates. Furthermore partial replacement of oxygen with nitrogen is readily obtained via ammonolysis.27–29 The anionic substitution of divalent oxygen for trivalent nitrogen additionally induces a change in the oxidation state of the transition metals involved. This oftentimes yields insulating materials as a consequence of strong perturbation of the periodic potential at the anion substructure and due to the increased covalency in the materials since N is less electronegative than O.30
A number of theoretical and experimental studies have focused on the electronic properties and electronic band gaps of the oxynitrides with spinel structure. Several studies have been carried out regarding ternary nitrides and oxynitrides31 in an effort to tailor the desirable mechanical and electronic properties associated with spinel oxynitrides. Research has expanded further into spinel-type oxynitrides in an effort to engineer the band gap value by varying the N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O ratio. However, the highly reducing atmosphere during thermal ammonolysis hardly permits middle-to-late transition metals to form oxynitrides without being reduced to elemental metals or metal nitrides. For this reason, the highest content of nitrogen reported in materials with spinel crystal structures is rather low.
:O ratio. However, the highly reducing atmosphere during thermal ammonolysis hardly permits middle-to-late transition metals to form oxynitrides without being reduced to elemental metals or metal nitrides. For this reason, the highest content of nitrogen reported in materials with spinel crystal structures is rather low.
In this study, we first doped N in the lattice of NiCr2O4 spinel by ammonolysis of the precursor oxides under favourable conditions. Subsequently we employed X-ray powder diffraction (XRD), magnetic susceptibility, X-ray photoelectron spectra (XPS) and heat capacity (Cv) measurements to investigate the structural and magnetic transitions in N doped NiCr2O4. We find this spinel exhibiting interesting physical properties: the Jahn–Teller (JT) distortion temperature decreases with increasing N concentration. In addition, the magnetic structure and magnetic interaction are also affected by the N doping.
2 Experimental details
Materials
NiCr2O4 is prepared by a solid state reaction of a stoichiometric mixture of powders of NiO (99.99%, Aldrich) and Cr2O3 (99.99%, Aldrich). The powders are ground together and heated for 24 h in air at a temperature of 1473 K with intermediate regrindings. 0.2–0.3 g of these samples are placed in an alumina boat. The boat is then placed in a silica tube which has air tight stainless steel end caps; these caps have welded valves and connections to input and output gas lines. All gases used are purified to remove trace amounts of oxygen or water using pellet copper, nickel, palladium and platinum with zeolites as support. The silica tube is then placed in a split tube furnace and the appropriate connections to gas sources are made. Argon gas is passed over the sample for 15 min to expel air before establishing a flow of ammonia gas (anhydrous, air gas). The NiCr2O4 is heated at the reaction conditions as summarized in Table 1. After treatment for the specified period, the furnace power is turned off and the product is cooled to room temperature for ∼4 h under ammonia flow conditions. Before the silica tube is taken out of the split tube furnace, argon gas is flowed through the silica tube to expel the ammonia gas. The samples treated at 773 K and 873 K are represented as N500 and N600, respectively.| Formula | NiCr2O4 | NiCr2O3.68N0.21 | NiCr2O3.55N0.30 |
| Formula weight | 226.68 | 224.50 | 223.69 |
| Crystal system | Tetrahedral | Cubic | Cubic |
| Space group | I41/amd | Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
Fdm |
| a/Å | 5.8319(1) | 8.3181(2) | 8.3184(1) |
| c/Å | 8.4433(1) | — | — |
| V/Å3 | 287.17(1) | 575.53(1) | 575.60(1) |
| ρcalcd/kg m−3 | 5.243 | 5.232 | 5.232 |
| wRp | 0.0462 | 0.0455 | 0.0410 |
| Rp | 0.0274 | 0.0263 | 0.0249 |
| Domain sizecalcd | 77 | 78 | 66 |
Characterization
Finely ground powders are examined with a Rigaku Ultima VI powder X-ray diffractometer (PXRD) with monochromatic CuKα1 radiation (λ = 1.5406 Å). Crystal structures of the oxide and the resulting nitride are confirmed by PXRD profiles using the GSAS package. Scanning electron microscopy (SEM) is performed with a LEO-1550 field emission SEM (FSEM). The dc-magnetic susceptibility is measured with a Physical Property Measurement System (PPMS) over the 4–350 K temperature range. Magnetization isotherms are measured at selected temperatures in the magnetic field range from −4 T to +4 T. Thermal gravimetric analyses (TGA) are performed with a SETSYS 16/18 (SETARAM) in a temperature range of 300 - 1273 K. The heat capacity measurement is performed using a Physical Property Measurement System (PPMS) in the temperature range from (1.9 to 400) K, and the measurement uncertainties are verified to be ±3% below 20 K and ±1% from (20 to 400) K.32 The detailed heat capacity measurement procedure can be found in the related work reported by Shi et al.32,33 The binding energy of the surface Ni, Cr, O, N species are determined by X-ray photoelectron spectra (XPS) with an ESCALAB250 X-ray photoelectron spectrometer using contaminated C as internal standard (C 1s = 284.6 eV).3 Results and discussion
The XRD patterns for the NiCr2O4 and the ammonolyzed products can be seen in Fig. 1. The X-ray diffraction profile is fitted to confirm the phase purity of the NiCr2O4 products as shown in Fig. S1.† The as prepared NiCr2O4 has a tetragonal structure in space group I41/amd with refined lattice parameters a = 5.8319(2) Å and c = 8.4433(2) Å (ref: Table 1). The ammonolysis product of NiCr2O4 at 873 K has a cubic structure in the space group Fdm with lattice parameters a = 8.3181(2) Å. The ammonolysis product of NiCr2O4 at 773 K is a mixture of two phases, containing mainly cubic phase. Presence of tetragonal NiCr2O4 in N500 is indicated by the small shoulder peak at ∼37 degrees in PXRD pattern. The slightly larger lattice parameters and volume for the oxynitride is due to the ionic radius of N3− (1.46 Å) being greater than the ionic radius of O2− (1.38 Å).34 Ammonolysis treatment of the NiCr2O4 at higher temperature (>873 K) results in decomposition to NiCrO3 and CrN. Both as-prepared and ammonolysis products of NiCr2O4 have similar average crystalline domain sizes of ∼76 nm, calculated from PXRD refinements. These results agreed well with results obtained from electron micrography. The precise location of the nitrogen atoms of the oxynitrides cannot be obtained by XRD due to the similar X-ray scattering factors of N3− and O2−. Hence measurement of neutron diffraction is needed; this will be pursued in future.
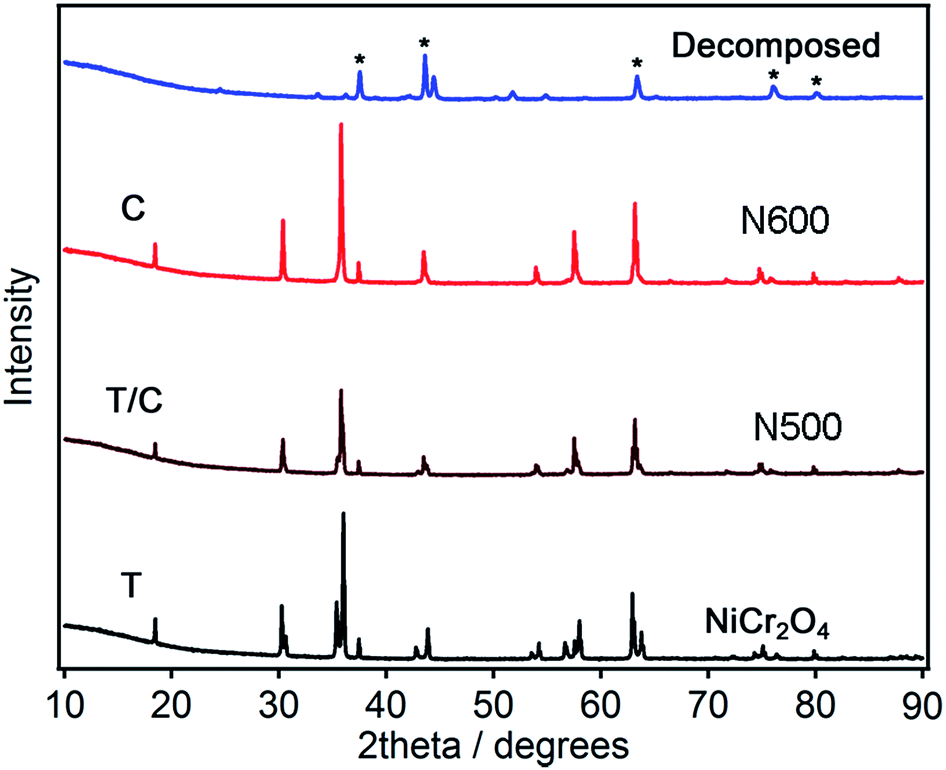 | ||
| Fig. 1 PXRD patterns for NiCr2O4: N500 and N600. T stands for tetrahedral phase and C stands for cubic phase of NiCr2O4. * stands for CrN. | ||
SEM images are used to observe the surface morphology of the products given in Fig. S2.† This figure confirms that the powder sample is contains large particle of size ∼70–400 nm. The ammonolyzed samples are designated as NiCr2O4−xN2x/3. The nitrogen contents are analyzed by converting NiCr2O4−xN2x/3 back to NiCr2O4 using TG in air flow as shown in Fig. S3.† The calculated average compositions are NiCr2O3.68N0.21 and NiCr2O3.55N0.30 for products ammonolysed at 773 and 873 K, respectively.
Fig. 2(A–D) presents the O 1s, N 1s, Cr 2p and Ni 2p core level X-ray photoelectron spectra (XPS) of N500 respectively. Each spectrum is fitted by a least squares method using Gaussian–Lorentzian envelopes. The surface stoichiometry of the sample calculated from the XPS data is N (3.73%), O (64.86%), Ni (10.98%), Cr (20.43%). As seen, the surface stoichiometry of the sample is close to the bulk of the sample; surface oxygen concentration however is quite different. The O 1s spectra are fitted with an O2− component at 529.9 eV along with additional peaks with binding energy (BE) at 530.9 eV and 531.9 eV. The most prominent peak at 529.9 eV corresponds to the lattice O2−, while the peak at 530.9 eV is assigned to the defect states within the crystal.35–38 The additional peak at 531.9 eV is associated with the O present in Cr–N–O or Ni–N–O bonds.39
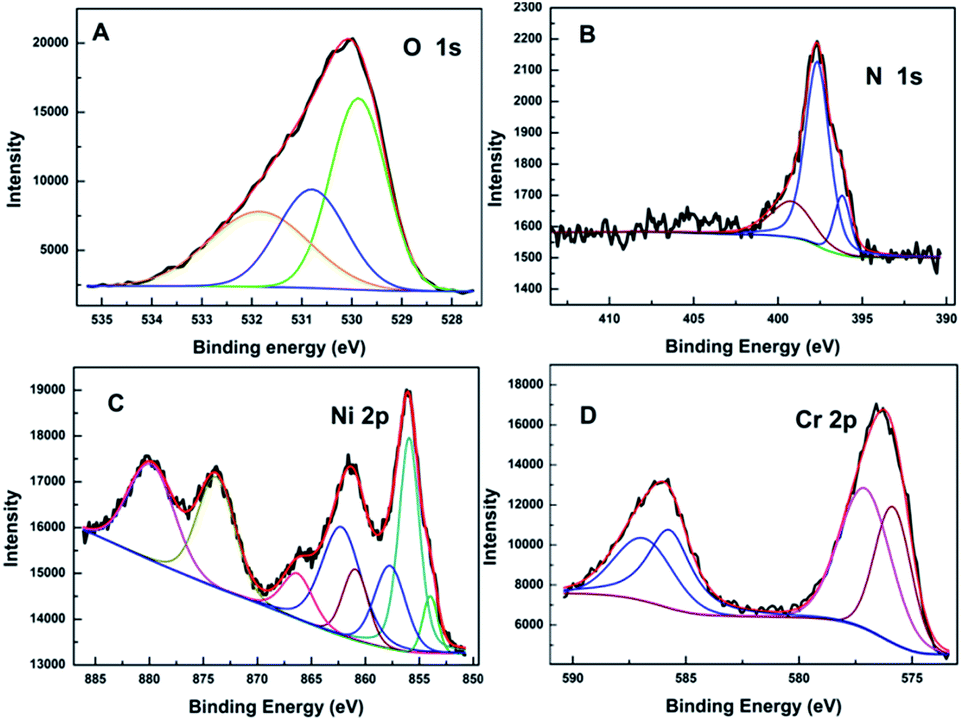 | ||
| Fig. 2 The O 1s (A), N 1s (B), Ni 2p (C) and Cr 2p (D) XPS spectra for N500. | ||
In Fig. 2(B), the N 1s features show only one asymmetric peak with a binding energy (BE) of 395–402 eV. On deconvolution, it can be seen that the broad peak consists of three different peaks at ∼396.2, 397.7 and 399.2 eV respectively. The main peak with the binding energies of 397.7 eV is assigned to Cr–N or Ni–N species;40,41 this suggests that nitrogen substitutes for oxygen in the spinel matrix. The peak with binding energy at ∼396.2 eV is indicative of a N3− species42,43 and confirms the presence of oxynitride species with lower binding energy.44 Interestingly the BE observed here for N3− is lower than that for NiN45 or CrN (396.3–397.1 eV);46,47 this suggests greater charge transfer to the nitrogen in NiCr2O4, when compared to the pure nitrides. However there could be a contribution from oxygen vacancy formation induced by N doping as well.48 The peak at 399.2 eV is attributed to nitrogen in the interstitial sites, where nitrogen has mixed Cr–O–N or O–Cr–N (Ni–O–N, O–Ni–N) environment. In this state, N atoms are bonded to one or more lattice oxygen ions47,49 and therefore are in a relatively positive oxidation state.50 From literature,18,21 the values of BE for the Ni 2p level and of the satellite shift ΔE permit identification of the Ni ions; they are divalent in tetrahedral environments. The Cr 2p spectra of the sample has a multiplet shape that is typical for Cr3+ ions.51 The incorporation of nitrogen into lattice results in Cr–N or Ni–N bonds, which is supported by the lower BE of Ni 2p and Cr 2p in N doped NiCr2O4. Marginal decrease in BE of Cr and Ni are reasonable since addition of N reduces the overall ionic character of the compounds, increasing the electron density around cations. Hence, in Fig. 2(C) and (D), the negative shifts are observed in Cr 2p and Ni 2p binding energies. The peak of Cr 2p is close to 576.0 eV which corresponds to Cr2+. Hence it is difficult to prove the existence of Cr2+ ions because of the small difference between the obtained value of binding energy and that of Cr2+. Therefore it is very likely that Cr ions exist in mixed oxidation states (i.e. 3+ as well as 2+) in N doped NiCr2O4.
To investigate the magnetic and structural evolution of the NiCr2O4 samples during the ammonolysis process in thermodynamics, the heat capacities of the NiCr2O4 and N doped samples are measured using the PPMS in the temperature range from (1.9 to 400) K. The experimental heat capacities are plotted against the temperature and compared with the reported values from Klemme et al.17 (Fig. 3). It can be seen from the figure that the heat capacity of the as-prepared NiCr2O4 are in agreement with published values revealing three thermal anomalies at around 310 K, 66 K and 29 K, respectively. Based on the previous magnetic and structure studies,13 the thermal anomaly appearing at 310 K (the first transition) has been attributed to a cooperative Jahn–Teller distortion lifting the orbital degeneracy in NiCr2O4, resulting in a structural transition from cubic to tetragonal. The other two anomalies at 66 K (the second transition) and 29 K (the third transition) have been accounted for by a longitudinal and a transverse antiferromagnetic ordering, respectively. As for the nitride samples; with increase in N content, the first and third transitions move to lower temperatures, while the second one moves to higher temperatures. The lattice heat capacities of these samples have been estimated by fitting the experimental heat capacities in the corresponding temperature windows to a combination of Debye and Einstein functions, and the fitting equation can be expressed by
| Cp,m = mD(ΘD/T) + n1E(ΘE,1/T) + n2E(ΘE,2/T) | (1) |
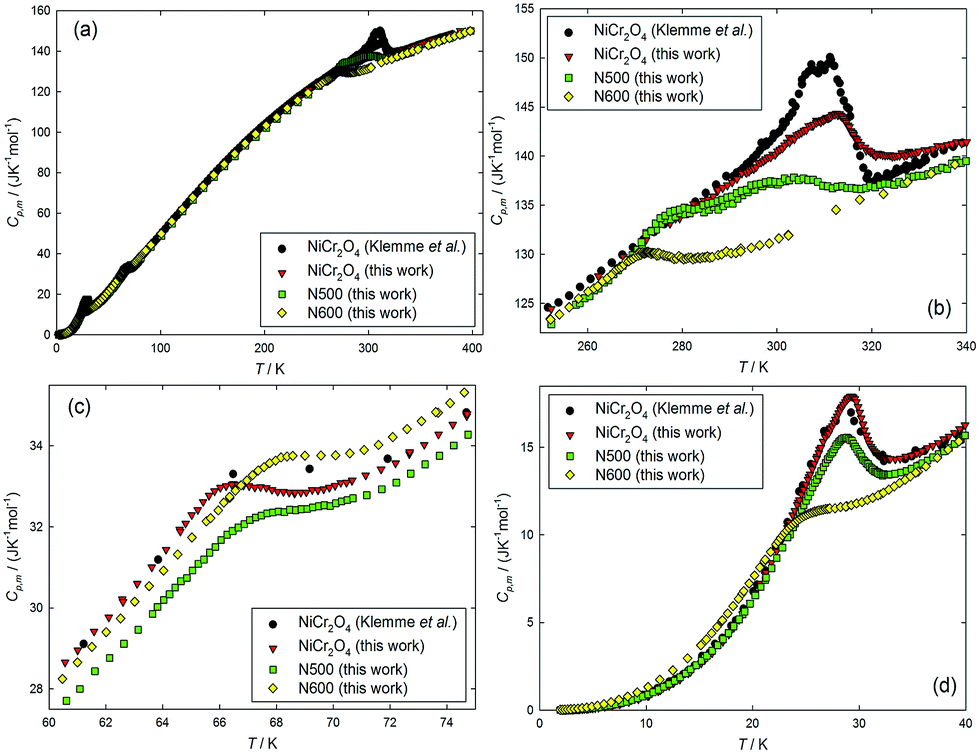 | ||
| Fig. 3 Plot of heat capacities of NiCr2O4 (measured by Klemme et al.17 and in this work), N500 and N600 as a function of temperature over the temperature ranges: (a) from (1.9 to 400) K, (b) from (250 to 340) K, (c) from (60 to 75) and (d) from (1.9 to 40). | ||
As can be seen from Fig. S4(a),† the peak temperature of the first transition moves to the lower temperatures with increase in N concentration. Also, the transition entropy decreases from 1.14 J K−1 mol−1 to 0.59 J K−1 mol−1 as the N anions concentration increases from NiCr2O4 to N600. This indicates that the Jahn–Teller distortion effect on the structural transition has been suppressed in these samples. The second transition due to the paramagnetic to ferrimagnetic ordering only has a slight influence on transition temperatures and entropies, suggesting that the doped N hardly affects the magnetic ordering. However, as for the third transition related to the antiferromagnetic ordering, the peak temperature moves from 29 K to 24.3 K, and the transition entropy changes to almost half the amount, decreasing from 2.53 J K−1 mol−1 to 1.30 J K−1 mol−1. This suggests that the doped N is likely to produce a significant effect on the thermodynamic behaviour of the third transition.
The heat capacity of a substance can be generally expressed as a sum of contributions from various modes related to the lattice, electronic, magnetic and others. Each of these contributions may be fitted to the corresponding theoretical models which would help in elucidation of important information about the physical properties of the substance.55 The lattice heat capacity far outweighs all other contributions at high temperature regions (above 15–20 K). However, at low temperatures (below 15–20 K) the lattice is comparable to the other contributions, and therefore each of these contributions can be extracted by fitting the total heat capacity to theoretical models in carefully chosen temperature regions. In order to further understand the observed specific heat capacity trends measured for NiCr2O4 and N-doped samples, the experimental heat capacities of these compounds below 10 K have been fitted to a theoretical function of:
| Cp,m = γT + B3T3 + B5T5 + B7T7 + BaswT3e−Δ/T | (2) |
Here the linear term is usually a contribution from electrons as well as vacancies or dislocations existing in the samples. The odd-powers in temperature represent the lattice vibration contribution. As for the magnetic contribution, it is represented using the term BaswT3e−Δ/T, where Basw is defined as the antiferromagnetic constant proportional to molar volume and the spin-wave stiffness constant. Δ is the magnetic spin-wave gap written in Kelvins.52–54 The antiferromagnetic contribution decreases in the N-doped samples, implying that the magnetic ordering is likely reduced to some extent as the nitrogen atoms get implanted in the lattice. Moreover the linear term tends to increase as we go from NiCr2O4 to N600, suggesting that the increasing nitrogen doping does result in increasing vacancy concentrations. The values relevant to the specific heat capacity measurements are listed in Table S3.† It can be seen from the results that the antiferromagnetic contribution does exist in the heat capacities. This further confirms the presence of antiferromagnetic ordering.
Fig. 4(a–c) shows the temperature dependence of susceptibility in NiCr2O4 and N doped samples, as measured after field cooling; a magnetic fields up to 1000 Oe is applied for getting the data. The observed magnetic data is analyzed using the equation given below:
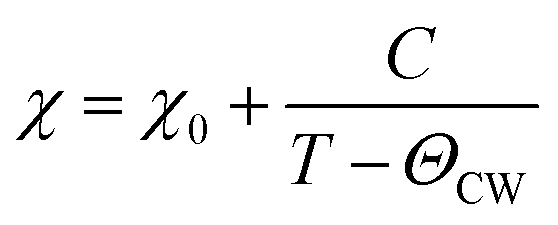 | (3) |
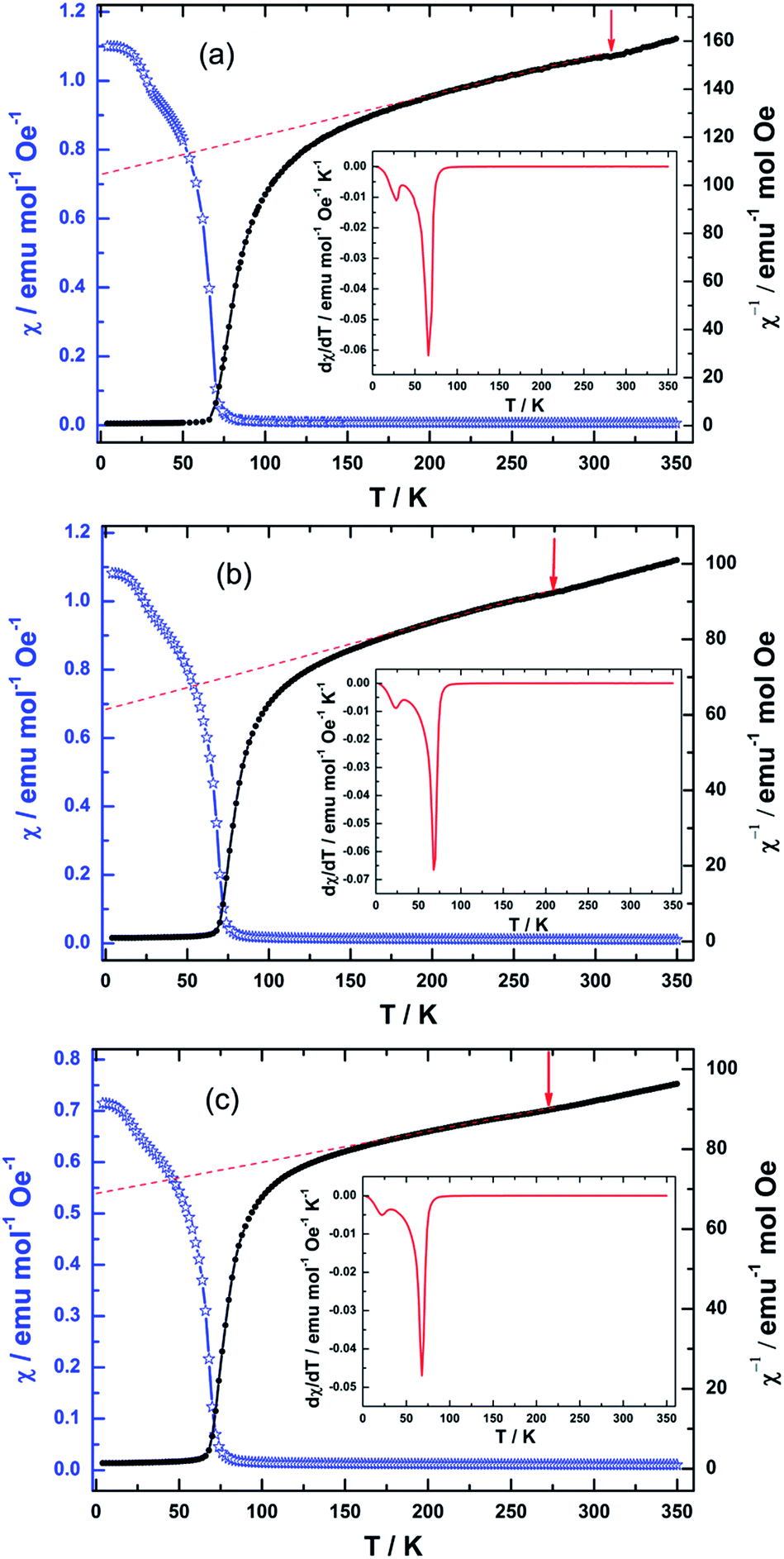 | ||
| Fig. 4 The temperature dependence of susceptibility (Blue) and inverse susceptibility curves (Black) for NiCr2O4 (a), N500 (b) and N600 (c). The arrows indicate the J–T transitions. The insets are the related temperature derivative of the susceptibilities, and the peaks corresponding to the TC and TS respectively. | ||
For the N doped samples, the J–T distortions occur at 275 K (N500) and 268 K (N600) respectively, which become lower than the transition temperature of pure NiCr2O4. The ferrimagnetic ordering (TC) occurs at 67 and 68 K, which is a minor increase after the N doping. In contrast to ferrimagnetic transition, the antiferromagnetic (transverse) ordering transition (TS) is decreasing from 28 K to 22 K with N doping. These transition temperatures corroborate well with the heat capacities results. When N atoms replace O atoms, the N ions would provide empty 2p orbitals for the formation of covalent bonds with Ni2+ and Cr3+.57 This likely causes a reduction of the J–T potential energy; the driving force for the J–T distortion would hence be reduced.58,59 The decreasing of distortion would enhance the contribution of the angular momentum and the spin–orbit coupling, and as a result, J–T transition temperature would further fall off.60 This in turn would stabilizes the cubic structure and leads to the gradual decrease in the tetragonal distortion temperature.
The Curie Weiss (CW) eqn (3) is applied to fit the paramagnetic regimes of the susceptibility before the onset of the J–T distortion for the NiCr2O4 and N doped samples. The obtained effective moments (μeff) and CW temperatures are listed in Table S4.† The experimental value of μeff is 6.43 μB for NiCr2O4, which is slightly larger than the expected 6.16 μB per formula unit.16 Hence we infer that although the angular momentum is largely quenched due to the tetragonal distortion, there is still a small contribution of the partially quenched orbital moments of Ni ions to the overall magnetic moment per formula unit. For the N doped NiCr2O4, the temperature independent Pauli term χ0 increases due to the covalent effect. The effective moments increase with increasing N content from 7.64 μB to 8.04 μB. This further corroborates the hypothesis that unquenched orbital moments of Ni2+ in the tetrahedral sites of the cubic structure make an important contribution to the magnetic moment.61 Magnetic contributions may also come from the impurity/dopant atoms,62 as well as the oxygen vacancies.63
The CW temperature of NiCr2O4 is −489 K, while N600 present even larger CW temperatures of −594 K. The exchange coupling between magnetic ions (Ni2+ and Cr3+) is directly proportional to CW temperature and can be expressed as:
| J = A|ΘCW| | (4) |
It should also be mentioned that the deviation of the reverse susceptibility against T curve from a straight line begins below a temperature of 176 K, 160 K, and 152 K respectively. The deviation observed is a slow downward drop. This downward deviation from the Curie–Weiss law indicates the onset of a magnetic short-range interaction just above TC. The decrease of deviation temperature means that N doping produces a decrease in correlation length.
To gain further insight into the magnetic properties of the synthesized N doped NiCr2O4 samples, magnetization (M) as a function of applied magnetic field (H) is measured at different temperatures (Fig. 5). For N500 and N600, the magnetization linearly increases with field while measured at 320 K and 100 K, and there is no magnetic hysteresis and hence the remanence (Mr) and coercivity (HC) are practically zero. This indicates a paramagnetic behavior. This is consistent with the M–T plots in Fig. 4. However, very clear magnetic hysteresis loops appear at 50 K with HC, Mr and spontaneous magnetization (MS) values listed in Table 2. The area under hysteresis increases with significant increase in coercivity (HC). We observe higher HC of 14 kOe for N600 compared with 8.3 kOe for NiCr2O4.10 The larger coercive field indicates presence of ferromagnetic long-range order and stronger magnetic anisotropy which may be caused by the spin–orbit interaction. At 50 K, the magnetization readily increases at low field and then linearly increases up to 40 kOe without showing any saturation for both N doped samples.
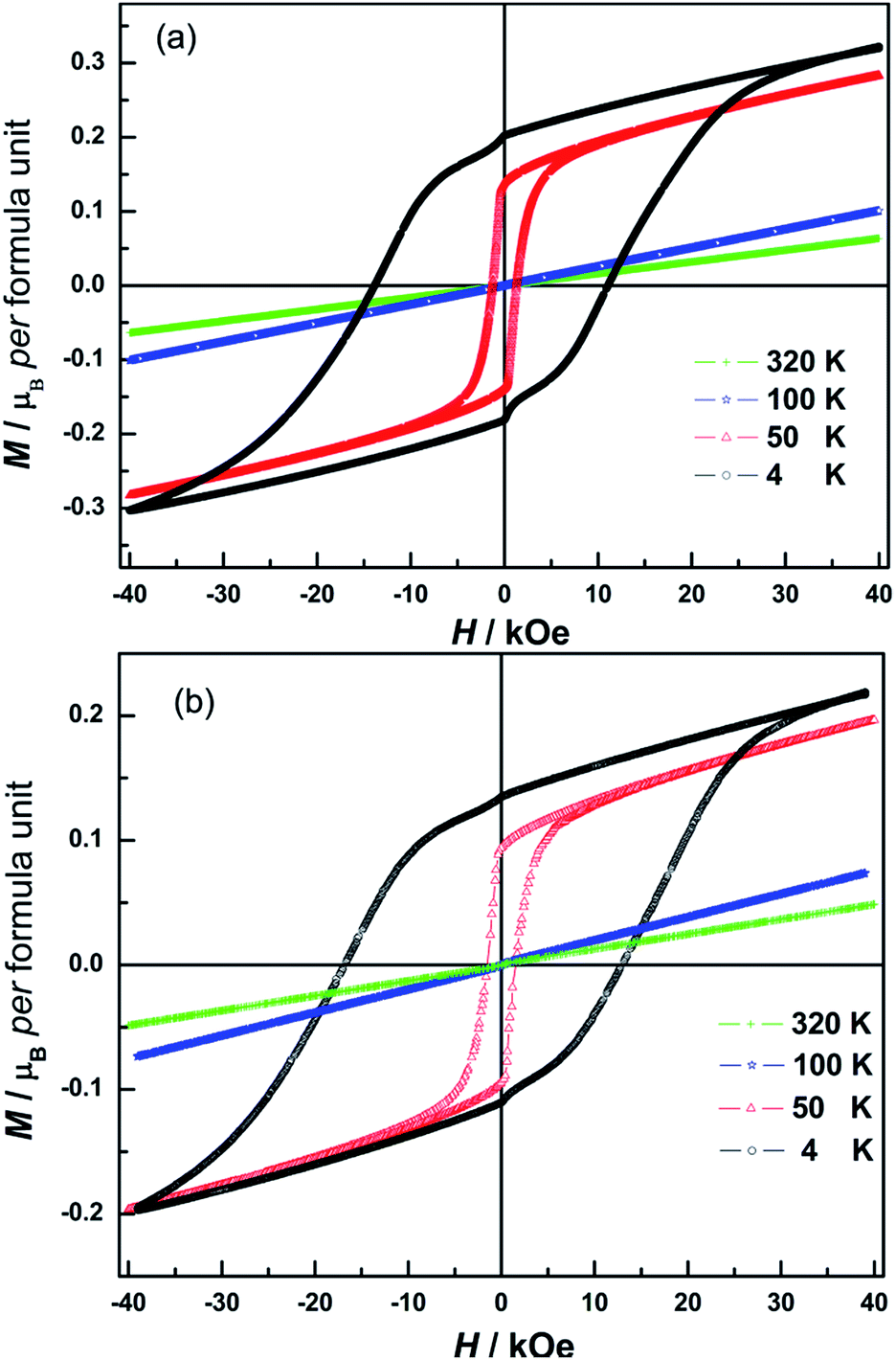 | ||
| Fig. 5 Isothermal field-dependent magnetization of N500 (a) and N600 (b) measured at 4 K, 50 K, 100 K and 320 K respectively. | ||
| Sample | C (emu K mol−1) | ΘCW (K) | Meff (μB) | χ0 (emu mol−1 Oe−1) | TS (K) | TN (K) | MS (μB) | HC (Oe) |
|---|---|---|---|---|---|---|---|---|
| NiCr2O4 | 5.18 | −486 | 6.43 | 0.00008 | 28 | 66 | 0.30 | 8300 |
| N500 | 7.30 | −456 | 7.64 | 0.00084 | 23 | 67 | 0.30 | 12500 |
| N600 | 8.08 | −594 | 8.04 | 0.00188 | 22 | 68 | 0.28 | 15000 |
When temperature goes down to 4 K, the isotherm M(H) still did not exhibit saturation, and the spontaneous magnetization of N doped NiCr2O4 is estimated to be 0.2 μB per formula unit. Considering the fact that HC is found to be less than 15 kOe, we suggest that this linear contribution signals a non-collinear spins arrangement of the NiCr2O4 ferrimagnet. As is stated in ref. 66 and 67, in non-collinear configuration the applied magnetic field exerts a torque that could change the angles between the canted magnetic moments. Hence the magnetization is expected to increase linearly with the magnetic field in this temperature regime. Since the magnetic moment of the tetrahedral sublattices in NiCr2O4 is greater in magnitude than that of the octahedral sublattice (MA > MB),68 and the magnetic structure consists of longitudinal and transverse magnetic sublattices with slight canting of spins.13 This suggests that when N is incorporated in the lattice, crystalline anisotropy and magnetostrictive effects associated with spin–orbit coupling have increased the noncollinearity of tetrahedral sublattice spins. This decreases the noncollinearity of octahedral sublattices.68 Hence the magnetic ordering in NiCr2O4 and the N doped samples is strongly coupled to structure. Further investigations are required to obtain greater clarity regarding correlations between magnetic and structural changes in this interesting material.
4 Conclusions
The N doped NiCr2O4 has been synthesized successfully by nitridation of the solid NiCr2O4 oxides at 773 K (N500) and 873 K (N600) respectively. The incorporation of N into the lattice has been confirmed using XRD and XPS measurements. The susceptibility and heat capacity based investigations indicate that structural and magnetic properties undergo significant changes upon N doping. Pure NiCr2O4 exists in tetragonal structure, however N500 and N600 contains cubic phase at room temperature. The temperature of cooperative Jahn–Teller distortion decreases with increase in N content. This observation is related to change in nature of bonding brought about by introduction on N into the lattice. The ferrimagnetic ordering temperature (TC) of the material exhibits a minor increase indicating that the A–B superexchange interaction is not affected much by the N-doping due to the empty eg orbital of Cr ions (which is present in the octahedral site). The N doped NiCr2O4 exhibits a canted ferrimagnetic structural transition between 30 and 70 K. We also report evidence for increased frustration and lowered correlation length with N doping in NiCr2O4. The hitherto unexplored details of magnetostructural coupling are likely to stimulate further investigations on this interesting material.Acknowledgements
This work is supported by National Natural Science Foundation of China through grant 11205160, 21471147, 21473198, and Liaoning Provincial Natural Science Foundation through grant 2014020087. Q. Shi would like to thank Hundred-Talent Program founded by Chinese Academy of Sciences. M. Yang would like to thank the National “Thousand Youth Talents” program of China. Tiju Thomas thanks the Department of Science and Technology, Government of India for Financial Support through INSPIRE Faculty Award (DST 01117), and for the Fast Track Young Scientist Award.References
- A. E. Ringwood, McGraw-Hill International Series in the Earth and Planetary Sciences: Composition and Petrology of the Earth's Mantle, 1975 Search PubMed.
- F. C. Romeijn, Philips Res. Rep., 1953, 8, 304–320 CAS.
- N. J. Jebarathinam, M. Eswaramoorthy and V. Krishnasamy, Bull. Chem. Soc. Jpn., 1994, 67, 3334 CrossRef CAS.
- G. Vivekanandan and V. Krishnasamy, Hung. J. Ind. Chem., 1995, 23, 21 CAS.
- J. Sloczynski, J. Ziolkowski, B. Grzybowska, R. Grabowski, D. Jachewicz, K. Wcislo and L. Gengembre, J. Catal., 1999, 187, 410 CrossRef CAS.
- B. L. Dubey, N. B. Singh, J. N. Srivastava and A. K. Ojha, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2001, 40, 841 Search PubMed.
- C. L. Honeybourne and R. K. Rasheed, J. Mater. Chem., 1996, 6, 277 RSC.
- N. Mufti, A. A. Nugroho, G. R. Blake and T. T. M. Palstra, J. Phys.: Condens. Matter, 2010, 22, 075902 CrossRef CAS PubMed.
- C. O. Augustion, D. Prabhakaran and L. K. Srinivasan, J. Mater. Sci. Lett., 1993, 12, 383 CrossRef CAS.
- T. D. Sparks, M. C. Kemei, P. T. Barton, R. Seshadri, E.-D. Mun and V. S. Zapf, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 024405 CrossRef.
- J. D. Dunitz and L. E. Orgel, J. Phys. Chem. Solids, 1957, 3, 20 CrossRef CAS.
- G. Ueno, S. Sato and Y. Kino, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1963 Search PubMed.
- K. Tomiyasu and I. Kagomiya, J. Phys. Soc. Jpn., 2004, 73, 2539 CrossRef CAS.
- E. Prince, J. Appl. Phys., 1961, 32, 68 CrossRef CAS.
- H. Ishibashi and T. Yasumi, J. Magn. Magn. Mater., 2007, 310, e610 CrossRef CAS.
- M. R. Suchomel, D. P. Shoemaker, L. Ribaud, M. C. Kemei and R. Seshadri, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 054406 CrossRef.
- S. Klemme and J. C. van Miltenburg, Phys. Chem. Miner., 2002, 29, 663 CrossRef CAS.
- M. Lenglet, A. D'Huysser, J. Arsene, J. P. Bonnelle and C. K. Joergensen, J. Phys. C: Solid State Phys., 1986, 19, L363 CrossRef CAS.
- M. Tovar, R. Torabi, C. Welker and F. Fleischer, Phys. B, 2006, 385–386, 196 CrossRef CAS.
- A. S. Mikheykin, D. Y. Chernyshov, A. A. Bush, A. S. Prokhorov, Y. I. Yuzyuk and V. P. Dmitriev, Phys. Solid State, 2014, 56, 785 CrossRef CAS.
- A. G. Kochur, A. T. Kozakov, K. A. Googlev, A. S. Mikheykin, V. I. Torgashev, A. A. Bush and A. V. Nikolskii, J. Electron Spectrosc. Relat. Phenom., 2014, 195, 208 CrossRef CAS.
- M. Ptak, M. Maczka, A. Pikul, P. E. Tomaszewski and J. Hanuza, J. Solid State Chem., 2014, 212, 218 CrossRef CAS.
- B. D. Hosterman, J. W. Farley and A. L. Johnson, J. Phys. Chem. Solids, 2013, 74, 985 CrossRef CAS.
- L. G. Antoshina, A. N. Goryaga and D. A. Chursin, Phys. Solid State, 2002, 44, 747 CrossRef CAS.
- B. L. Morris, P. Russo and A. Wold, J. Phys. Chem. Solids, 1970, 31, 635 CrossRef CAS.
- A. V. Powell, D. C. Colgan and C. Ritter, J. Solid State Chem., 1997, 134, 110 CrossRef CAS.
- M. J. Martinez-Lope, M. T. Casais and J. A. Alonso, Z. Naturforsch., B: J. Chem. Sci., 2006, 61, 164 CAS.
- M. T. Weller and S. J. Skinner, Int. J. Inorg. Mater., 2000, 2, 463 CrossRef CAS.
- M. Retuerto, C. de la Calle, M. J. Martinez-Lope, F. Porcher, K. Krezhov, N. Menendez and J. A. Alonso, J. Solid State Chem., 2012, 185, 18 CrossRef CAS.
- G. M. Veith, M. Greenblatt, M. Croft and J. B. Goodenough, Mater. Res. Bull., 2001, 36, 1521 CrossRef CAS.
- T. D. Boyko, C. E. Zvoriste, I. Kinski, R. Riedel, S. Hering, H. Huppertz and A. Moewes, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 085203 CrossRef.
- R. Dai, S. Zhang, N. Yin, Z.-C. Tan and Q. Shi, J. Chem. Thermodyn., 2016, 92, 60 CrossRef CAS.
- Q. Shi, C. L. Snow, J. Boerio-Goates and B. F. Woodfield, J. Chem. Thermodyn., 2010, 42, 1107 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- H. A. E. Hagelin-Weaver, J. F. Weaver, G. B. Hoflund and G. N. Salaita, J. Electron Spectrosc. Relat. Phenom., 2004, 134, 139 CrossRef CAS.
- P. R. Norton, R. L. Tapping and J. W. Goodale, Surf. Sci., 1977, 65, 13 CrossRef CAS.
- B. P. Payne, M. C. Biesinger and N. S. McIntyre, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 159 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. Smart, Appl. Surf. Sci., 2011, 257, 2717 CrossRef CAS.
- X. B. Chen and C. Burda, J. Phys. Chem. B, 2004, 108, 15446 CrossRef CAS.
- S. Agouram, F. Bodart and G. Terwagne, J. Electron Spectrosc. Relat. Phenom., 2004, 134, 173 CrossRef CAS.
- I. Milosev, H. H. Strehblow and B. Navinsek, Surf. Coat. Technol., 1995, 74–75, 897 CrossRef CAS.
- M. Batzill, E. H. Morales and U. Diebold, Phys. Rev. Lett., 2006, 96, 026103 CrossRef PubMed.
- S. Q. Xiao and O. Takai, Thin Solid Films, 1998, 317, 137 CrossRef CAS.
- I. Milosev, J. M. Abels, H. H. Strehblow, B. Navinsek and M. Metikos-Hukovic, J. Vac. Sci. Technol., A, 1996, 14, 2527 CAS.
- G. Soto, W. de la Cruz and M. H. Farias, J. Electron Spectrosc. Relat. Phenom., 2004, 135, 27 CrossRef CAS.
- C. Palacio, A. Arranz and D. Diaz, Thin Solid Films, 2006, 513, 175 CrossRef CAS.
- B. Subramanian and M. Jayachandran, Corros. Eng., Sci. Technol., 2011, 46, 554 CrossRef CAS.
- S.-H. Lee, E. Yamasue, K. N. Ishihara and H. Okumura, Appl. Catal., B, 2010, 93, 217 CrossRef CAS.
- Y. Cong, J. Zhang, F. Chen and M. Anpo, J. Phys. Chem. C, 2007, 111, 6976 CAS.
- R. A. R. Monteiro, S. M. Miranda, V. J. P. Vilar, L. M. Pastrana-Martinez, P. B. Tavares, R. A. R. Boaventura, J. L. Faria, E. Pinto and A. M. T. Silva, Appl. Catal., B, 2015, 162, 66 CrossRef CAS.
- M. C. Biesinger, C. Brown, J. R. Mycroft, R. D. Davidson and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1550 CrossRef CAS.
- Q. Shi, L. Zhang, M. E. Schlesinger, J. Boerio-Goates and B. F. Woodfield, J. Chem. Thermodyn., 2013, 62, 35 CrossRef CAS.
- Q. Shi, L. Y. Zhang, M. E. Schlesinger, J. Boerio-Goates and B. F. Woodfield, J. Chem. Thermodyn., 2013, 62, 86 CrossRef CAS.
- Q. Shi, L. Y. Zhang, M. E. Schlesinger, J. Boerio-Goates and B. F. Woodfield, J. Chem. Thermodyn., 2013, 61, 51 CrossRef CAS.
- E. S. R. Gopal, Specific heats at low temperatures, The International Cryogenics Monograph Series, 1966 Search PubMed.
- O. Crottaz, F. Kubel and H. Schmid, J. Mater. Chem., 1997, 7, 143 RSC.
- M. H. Whangbo, H. J. Koo, A. Villesuzanne and M. Pouchard, Inorg. Chem., 2002, 41, 1920 CrossRef CAS PubMed.
- J. B. Goodenough and A. L. Loeb, Phys. Rev., 1955, 98, 391 CrossRef CAS.
- L. Cianchi, M. Mancini and P. Moretti, Phys. Rev. B: Solid State, 1973, 7, 5014 CrossRef CAS.
- J. B. Goodenough, Magnetism and the chemical bond, 1963 Search PubMed.
- J. S. Smart, Phys. Rev., 1954, 94, 847 CrossRef CAS.
- A. Stashans and S. Jacome, Comput. Mater. Sci., 2014, 81, 353 CrossRef CAS.
- A. Sundaresan, R. Bhargavi, N. Rangarajan, U. Siddesh and C. N. R. Rao, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 161306 CrossRef.
- D. G. Wickham and J. B. Goodenough, Phys. Rev., 1959, 115, 1156–1158 CrossRef CAS.
- S. Blanco-Canosa, F. Rivadulla, V. Pardo, D. Baldomir, J. S. Zhou, M. Garcia-Hernandez, M. A. Lopez-Quintela, J. Rivas and J. B. Goodenough, Phys. Rev. Lett., 2007, 99, 187201 CrossRef CAS PubMed.
- D. Tobia, J. Milano, M. T. Causa and E. L. Winkler, J. Phys.: Condens. Matter, 2015, 27, 016003 CrossRef PubMed.
- I. S. Jacobs, J. Phys. Chem. Solids, 1960, 15, 54 CrossRef CAS.
- A. N. Goryaga, L. G. Antoshina, A. I. Kokorev and D. A. Chursin, Phys. Solid State, 2002, 44, 759 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra22773b |
| This journal is © The Royal Society of Chemistry 2016 |