
Dual-functionalisation of gelatine nanoparticles with an anticancer platinum(ii)–bisphosphonate complex and mineral-binding alendronate†

Abstract
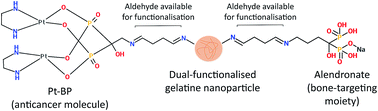
In order to improve the efficacy of therapeutic systems to treat bone tumours, novel drug delivery vehicles should be developed that have strong and specific affinity to mineralised tissue and at the same time are able to release anticancer molecules locally in a controlled and sustained manner. Recently, we developed mineral-binding gelatine nanoparticles with enhanced affinity to calcium phosphate by conjugating alendronate (ALN) molecules onto their surface. Herein, we have enhanced the functionality of these nanoparticles by rendering them potentially therapeutically active via covalent linking of an anticancer platinum–bisphosphonate (Pt–BP) complex. Different functionalisation schemes and molar ratios between reactants were screened and the effective functionalisation of gelatine nanoparticles with Pt–BP (or with both Pt–BP and ALN) was assessed. Our results revealed that the degree of functionalisation could be tailored by varying the molar ratio of Pt–BP and ALN relative to glutaraldehyde used as crosslinker. A sustained and tunable release of platinum as a function of the initial Pt–BP/ALN/glutaraldehyde molar ratio was achieved for both Pt–BP- and dual-functionalised gelatine nanoparticles. Moreover, dual-functionalised gelatine nanoparticles also displayed a high affinity to hydroxyapatite-coated surfaces thanks to the presence of ALN. Summarising, it was demonstrated that mineral-binding gelatine nanoparticles can be loaded with tailored amounts of anticancer molecules, which may benefit the development of bone-seeking carriers for targeted delivery of drugs to treat bone tumours.
 Please wait while we load your content...
Please wait while we load your content...

