
An investigation on the aqueous-phase hydrodeoxygenation of various methoxy-substituted lignin monomers on Pd/C and HZSM-5 catalysts†
Cong Zhang‡
a,
Jingbo Qi‡a,
Jing Xingab,
Si-Fu Tanga,
Liang Song*a,
Yuanyuan Suna,
Chuanhui Zhanga,
Hongchuan Xina and
Xuebing Li*a
aKey Laboratory of Biofuels, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao, 266101, China. E-mail: lixb@qibebt.ac.cn
bSchool of Chemistry and Materials Science, Liaoning Shihua University, Fushun, 113001, China
First published on 21st October 2016
Abstract
Aqueous phase catalytic upgrading of lignin monomers to hydrocarbons via hydrodeoxygenation (HDO) has been explored using a combination of Pd/C and HZSM-5 catalysts under 2 MPa of H2 (ambient temperature). Model monomers with varying numbers of methoxy groups, including phenol, anisole, guaiacol and 2,6-dimethoxy-phenol, were chosen as lignin model compounds. Mechanistic studies revealed cascade and parallel reaction pathways via hydrogenation and dehydration (hydrolysis) processes, which were catalyzed by Pd/C and HZSM-5, respectively. Hydrogenation was preferred at lower temperature, whereas higher temperature was favorable for the removal of oxygen-containing functional groups. The effect of methoxy groups on the HDO of these monomers was also investigated systematically. Basically, the conversion of multi-substituted monomers was tougher than that of mono-substituted ones, due to steric constraint and the inhibition of the electron-donating hydroxyl group. The selectivities to cyclohexane from phenol and anisole were improved significantly by increasing the temperature to 413 K. However, cyclohexanone was preferably produced over cyclohexane when using model compounds with multi-substituents (guaiacol and 2,6-dimethoxy-phenol), even at temperatures above 513 K. Comparative experiments were also conducted on the HDO of 1,2-cyclohexanediol with or without the presence of phenol, which clearly suggested that the further HDO of guaiacol and 2,6-dimethoxy-phenol was inhibited, probably due to the strong interactions between cyclohexanediol molecules and Brønsted acid sites.
1. Introduction
Currently, plant biomass represents a sustainable organic carbon source,1–3 and biofuel derived from plant biomass is by far the most unique sustainable type of liquid fuel. Compared to fossil fuels, levels of greenhouse gas released by biofuels are significantly lower, and biofuels can even be carbon neutral if they are produced using efficient methods.4–6 However, the direct application of bio-oil is limited by its corrosivity, low vapor pressure, high viscosity and instability.7–9 Because biomass-derived platform molecules are generally highly oxygenated compounds (15–40 wt% of its dry mass),10 employing an upgrading process before their application is necessary to overcome the deleterious properties of biomass pyrolysis oil.11 Hydrodeoxygenation (HDO) processes have recently received considerable attention as the most effective method for upgrading bio-oil into liquid transportation fuels, through which the effective H/C ratio is improved.12 Use of a carefully designed catalyst is required in order to remove oxygen to the maximum degree with the minimum amount of hydrogen consumed (i.e. low H2 pressure), which is the key challenge of HDO.13Phenolic compounds (phenol, guaiacol and other substituted phenol compositions) derived from the decomposition of lignin8 are the least reactive chemical compounds found in biomass pyrolysis oils14 and represent a significant fraction of the total bio-oil15 and other biomass- or lignin-derived product streams.16,17 Therefore, it is very important to identify the reaction pathways that phenols may undergo.18–22 It has been proposed that aqueous-phase dehydration is the key step in the overall HDO reaction, and this is consistent with the high apparent activation energy.10,23 C–O bond cleavage is the rate-determining step in the conversion of β-O-4 and 4-O-5 ether bonds and can be catalyzed via hydrogenolysis using metal catalysts.24 Meanwhile, parallel hydrogenolysis and hydrolysis account for the cleavage of the C–O bond of the 4-O-5 linkage.25 As reported previously, the high selectivity of HDO in the conversion of lignin-derived phenolic monomers and dimers to cycloalkanes has been achieved on Pd/C and HZSM-5 catalysts in aqueous phase at 473 K.26 The reactivity of mono- and dimeric lignin model compounds has also been investigated on metal and acid co-catalysts. Dimeric oxygen-bridged model compounds such as benzylphenyl ether and diphenyl ether can be readily converted to monomeric species, suggesting that the reaction mechanisms of monomeric species serve as a basis for investigating the HDO of lignin model compounds.10 The reaction pathways and product distributions can be affected by various factors, such as the acidity of the zeolite,27 the sizes of the nanoparticles,28 the reaction temperature,29 and the Lewis basicity of the solvent.30 Additionally, the substitution of the phenyl group can also affect the HDO of monomeric phenol compounds. It has been reported that the HDO of diverse para-substituted phenolic monomers can be achieved effectively with satisfying conversion rates.31–33 However, the deoxygenation and ring hydrogenation of guaiacol on Pt-modified zeolites can both be significantly suppressed by the methoxy group in comparison with phenol (no substitution) and o-cresol (methyl substitution), because the adsorption modes and transport parameters are affected by their molecular dimensions.34
Considering the ubiquity of methoxy groups in bio-oil, a systematic study on the effect of substituent methoxy groups on the HDO of lignin-derived monomeric phenols is urgently required. Very few studies have focused on this, even though there is increasing interest in biofuel production via bio-oil upgrading.35–39 Therefore, we decided to investigate the reaction pathways of various methoxy-substituted phenolic monomers. Phenol, anisole, guaiacol and 2,6-dimethoxy-phenol were selected as starting model compounds based on the following considerations: (1) phenol is an attractive model compound for investigating principal hydrogenation and deoxygenation routes; (2) anisole (or methoxy-benzene) can be used to explore the relative reactivity of the methoxy group; (3) guaiacol and 2,6-dimethoxy-phenol, possessing two types of C–O bonds, Csp2–OH (hydroxyl) and Csp2–OCH3 (methoxy), are good model compounds for probing the catalytic behavior with respect to these different organic functions.
2. Experimental section
2.1 Chemicals and commercial catalysts
The chemicals were obtained from commercial suppliers: phenol (Aladdin, ≥99.5% standard for GC), anisole (Aladdin, ≥99.5% standard for GC), cyclohexanol (Aladdin, ≥99.0% standard for GC), methoxy-cyclohexane (J&K, 98% GR assay), guaiacol (Aladdin, 99.0% AR assay), 2,6-dimethoxy-phenol (Aldrich, 99%), ethyl acetate (Aladdin, 99.5% GR assay), ZSM-5 (Catalyst Plant of Nankai University, NKF-5; Si/Al = 12.5), Pd/C (Aladdin, loading 5 wt% Pd).2.2 Catalyst preparation
The purchased catalysts were washed with deionized water at ambient temperature. After drying at 393 K overnight, the sample was calcined in air (flow rate: 30 mL min−1 g cat−1) at 823 K for 4 h with a heating rate of 3 K min−1 in order to remove the template. The catalysts were tested after Na-ZSM-5 was converted into the acidic form (H+) by refluxing it three times in 1 mol L−1 NH4NO3 at 313 K for 6 h. The solid was filtered, washed, dried at 393 K overnight and calcined at 823 K for 4 h (heating rate: 1 K min−1) under air flow (60 mL min−1). The samples in the NH4+-form were calcined at 823 K for 4 h (heating rate: 1 K min−1) to convert them into the H+-form.2.3 Catalytic measurements
The catalytic HDO reaction was performed in a stainless-steel autoclave with an internal volume of 80 mL. Typically, 0.0025 mol of phenolic model compound, 0.05 g of Pd/C and 0.5 g of HZSM-5 and 20 mL of water were added into the autoclave reactor. It was then flushed with H2 three times and pressurized with H2 to a total pressure of about 2 MPa at room temperature. The autoclave was then heated to a designated temperature for 2 h under stirring at 680 rpm. After the reaction, the reactor was cooled down to room temperature in an ice bath. The organic phase was extracted with ethylacetate (3 × 10 mL). The gas phase, organic phase and aqueous phase were separated and analyzed using a GC-MS (Agilent 7890A/5975C) equipped with a capillary column (HP-5; 30 m × 250 μm × 0.25 μm) and a flame ionization detector (FID). It was found that only hydrogen and trace amounts of CO2 and methanol existed in the gas phase. As a result, only the changes in the liquid phase were considered.The conversion and selectivity were calculated using carbon balance equations. Conversion = 100% − (C atoms in residual reactants/total C atoms in the products besides residual reactants) × 100%. Selectivity = (C atoms in each product/total C atoms in the products) × 100%. 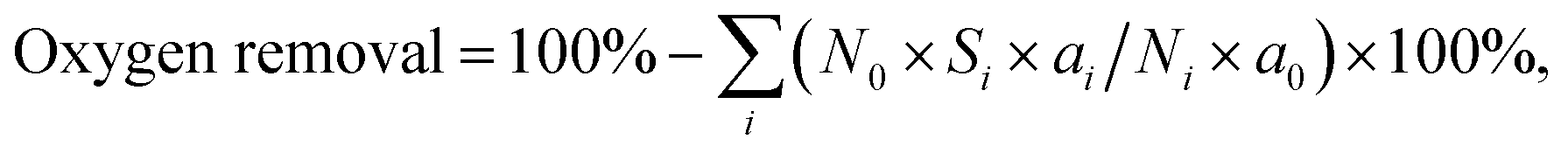 where N0 is the number of C atoms in a reactant molecule, Ni is the number of C atoms in the i-product molecule, Si is the selectivity of the i-product, a0 is the number of O atoms in the reactant molecule and ai is the number of O atoms in the i-product molecule.
where N0 is the number of C atoms in a reactant molecule, Ni is the number of C atoms in the i-product molecule, Si is the selectivity of the i-product, a0 is the number of O atoms in the reactant molecule and ai is the number of O atoms in the i-product molecule.
2.4 Catalyst characterization
The X-ray diffraction (XRD) pattern was collected on a Bruker D8 Advance diffractometer (Cu Kα = 1.54056 Å) at 40 kV/100 mA on a spinner in the range of 2θ = 5–50° and with a step size of 0.01°. Scanning electron microscopy (SEM) was performed on a Hitachi S-4800 SEM-microscope with an accelerating voltage of 5 kV. Prior to scanning, the dry samples were platinum-coated. Energy-dispersive X-ray (EDX) analysis was conducted on an Oxford Inca X-ray energy detector. Transmission electron micrographs (TEM) were recorded on a field emission H-7600 electron microscope operated at 120 kV. Before the measurement, the samples were prepared by depositing a drop of an ultrasonicated methanol suspension of the solid material onto a carbon-coated Cu grid for TEM measurement. Nitrogen adsorption–desorption experiments were carried out at 77 K using a Micromeritics ASAP 2000-M+C apparatus. Prior to adsorption, the sample was degassed in a vacuum at 523 K for 24 h. The specific surface area was determined according to the Brunauer–Emmett–Teller (BET) method in the linear range of p/p0 = 0.01–0.25. The t-plot was used to evaluate the external surface area and the micropore volume. The mesopore volume was then determined using the BJH method. As shown in Table S1,† the Si/Al ratio, BET surface areas, and amounts and properties of acid sites on the zeolite catalyst were characterized and calculated. As evidenced by the BJH mesopore size distributions, the sample ZSM-5 contained almost no mesopore volume (0.04 cm3 g−1) and a relatively small external surface area (ca. 13 m2 g−1).The acidic properties of HZSM-5 were characterized using NMR and NH3-TPD. The NMR experiment was conducted on a Bruker Ascend-500 spectrometer at resonance frequencies of 130.24 MHz for 27Al. 27Al NMR experiments were recorded on a 4 mm triple-resonance MAS probe at a spinning rate of 10 kHz. Pulse MHz 27Al NMR experiments were recorded on a 4 mm triple-resonance MAS probe at a spinning rate of 10 kHz. The pulse width (p/2) for 27Al was measured to be 1.7 μs. 27Al MAS NMR spectra were recorded using a small-flip-angle technique with a pulse length of 0.28 l s (p/12) and a recycle delay of 1 second. Temperature programmed desorption of NH3(NH3-TPD) over the HZSM-5 catalysts was performed on a Micromeritics 2920TR chemisorption analyzer. Firstly, around 15 mg of sample was activated by heating it at 773 K for 1 h in Ar with a heating rate of 10 K min−1. After cooling to 373 K, NH3 adsorption was carried out. NH3 was adsorbed for 2 h by adding 10 vol% to the Ar carrier gas with a total flow 40 mL min−1 at 373 K. Physically adsorbed NH3 was removed by degassing at 373 K for 2 h with Ar (20 mL min−1) until no further weight loss was observed. The NH3-TPD of the sample was carried out by heating the sample under flowing Ar at a rate of 10 K min−1 from 373 to 873 K and species desorption was detected using a TCD (thermal conductivity detector). To calibrate the method, a standard calibration experiment was performed.
In addition, the numbers of weak and strong acid sites were obtained by analyzing the amount of ammonia desorbed by integrating the TPD curves using the Gauss curve fitting method (Fig. S3†). It has previously been reported that the increase in the quantity of acid sites and the decrease in the activation energy of ammonia desorption on the strong acid sites of HZSM-5 is followed by a decrease in the Si/Al ratio.27 Consequently, Pd/C and HZSM-5 in a Si/Al ratio of 15 were jointly used as a catalyst in this work, due to their excellent performance in hydrodeoxygenation previously.27
3. Results and discussion
3.1 Characterization of the catalyst
The powder XRD pattern of HZSM-5 exhibits well-resolved diffraction peaks (see Fig. S1 in the ESI†), which are characteristic of the MFI framework structure. SEM images of the HZSM-5 catalyst and TEM images of Pd/C are shown in Fig. S1 and S2.† The HZSM-5 catalyst is irregularly shaped and demonstrates a laminated structure, but shows a uniform crystallite size in the range of 1.5–2 μm. Pd particles are uniformly supported on the carbon base, but the formation of aggregates is also observed.27Al MAS NMR was used to detect the coordination state of aluminum species in the HZSM-5 catalyst (Fig. S1†). Apparently, the sample exhibits a strong peak centered at 53 ppm and a small peak centered at 1 ppm, which are attributed to tetrahedrally coordinated aluminum (framework Al, FAl) and octahedrally coordinated aluminum (extra-framework Al, EFAl), respectively. This indicates that Al in the sample is mainly tetrahedrally coordinated.
3.2 HDO of mono-substituted lignin monomers
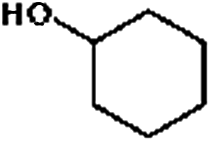
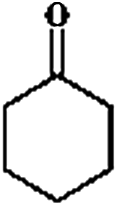
The HDO of phenol was conducted under acid-free conditions to clarify the role of Pd/C (Table 1). It was found that the aromatic ring was completely hydrogenated to cyclohexanol at 413 K, with 0.1% cyclohexanone as an intermediate and no cyclohexane detected. It is obvious that Pd/C is much more efficient in hydrogenation than in deoxygenation.10,40,41| Entry | Reaction temperature (K) | Selectivity of products (C%) |
|
|||
|---|---|---|---|---|---|---|
|
|
|
|
|||
| a Typical reaction conditions: phenol (0.005 mol), HZSM-5 (Si/Al = 15, 0.5 g), Pd/C (0.05 g), H2O (20 mL), 2 MPa H2, 2 h, stirred at 680 rpm.b Using only Pd/C as the catalyst. | ||||||
| 1 | 383 | 0.0 | 99.8 | 0.2 | 0.0 | 100.0 |
| 2 | 413 | 49.2 | 50.8 | 0.0 | 0.0 | 100.0 |
| 3 | 443 | 99.6 | 0.0 | 0.0 | 0.4 | 100.0 |
| 4 | 473 | 99.9 | 0.0 | 0.0 | 0.1 | 100.0 |
| 5 | 413b | 0.0 | 99.9 | 0.1 | 0.0 | 100.0 |

As proposed in ref. 37 and 42, the mechanism for gas-phase phenol conversion to cyclohexane using heterogeneous sulfide catalysts under 523 K and 100 bar of H2 pressure involves direct hydrogenolysis to benzene followed by benzene hydrogenation, or first hydrogenation to cyclohexanol followed by cyclohexanol hydrogenolysis. However, in this contribution using Pd/C and HZSM-5 in the aqueous-phase, the HDO of phenol and cyclohexanol observed did not follow the classical hydrogenolysis route under lower temperature and H2 pressure. The mechanism for the aqueous-phase HDO of phenol to cyclohexane using the Pd/C and acid catalysts proceeds via an initial metal-catalyzed aromatic ring hydrogenation, naphthenic alcohol dehydration, and subsequently metal-catalyzed cycloalkane hydrogenation (Scheme 1a). This is similar to the overall reaction pathway proposed previously using Pd/C and phosphoric acid as bi-functional catalysts.43
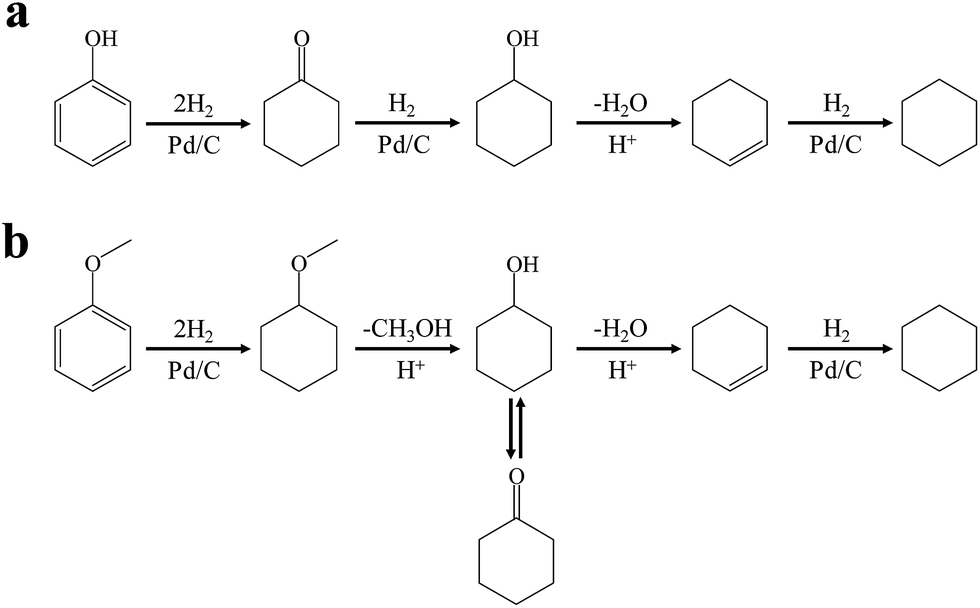 | ||
| Scheme 1 Reaction pathways for the conversion of phenolic compounds to cyclohexane over Pd/C and HZSM-5 in the liquid phase: (a) phenol; (b) anisole. | ||
The conversion and selectivity for phenol hydrogenation over Pd/C and HZSM-5 as a function of reaction temperature are shown in Table 1. When the reaction temperature was as low as 383 K, the amount of cyclohexane was below the detection limit and the product distribution was similar to that under acid-free conditions. The selectivity of cyclohexane via the formation of cyclohexanol from phenol was improved to 49.2% when the temperature was increased from 383 K to 413 K, and was even higher at 443 K and 473 K. The deoxygenation of the cyclohexanol intermediate was inhibited without the addition of HZSM-5 or under low temperature (383 K). In line with previous studies,10 the combination of acidic conditions and elevated reaction temperature resulted in the almost complete HDO of phenol to cyclohexane with approximately 100% selectivity.
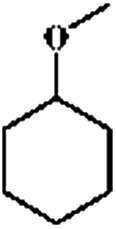
As a typical component of bio-oil, anisole (methyl phenyl ether or methoxy-benzene) contains a methoxy group that provides interesting chemistry. Bimolecular reactions including the following transalkylation reactions were observed by Zhu et al.:44 (a) anisoles to phenol and methyl-anisole; (b) phenol and methyl-anisole to cresols; (c) phenol and anisole to cresol and phenol; (d) methyl-anisole and cresol to phenol and xylenol. However, the hydrogenation of the aromatic ring and methoxy hydrolysis proceeds at the same time on the dual-functional catalyst system of Pd/C and H3PO4 under the selected conditions, while the hydrolysis of anisole to phenol is the dominating reaction.24 In our work, according to the conversion and product distribution of anisole over Pd/C and an HZSM-5 zeolite, as displayed in Table 2 at varying reaction temperatures, the mechanism varies significantly. The selectivity of methoxy-cyclohexane and cyclohexanone (intermediate) reaches 29.0 and 34.6% at 383 K, respectively. When the reaction temperature exceeds 413 K, the yield of cyclohexane is greatly improved to approximately 80%, with only trace amounts of methoxy-cyclohexane and cyclohexanone detected (Table 2). During anisole conversion, there is no phenol detected when the temperature is varied, indicating that the hydrogenation of the aromatic ring is the main reaction pathway (Scheme 1b).
| Entry | Reaction temperature (K) | Selectivity of products (C%) |
|
|||||
|---|---|---|---|---|---|---|---|---|
| CH3OH |
|
|
|
|
|
|||
| a Typical reaction conditions: anisole (0.005 mol), HZSM-5 (Si/Al = 15, 0.5 g), Pd/C (0.05 g), H2O (20 mL), 2 MPa H2, 2 h, stirred at 680 rpm. | ||||||||
| 1 | 383 | 10.1 | 0.3 | 29.0 | 25.9 | 34.6 | 0.0 | 92.4 |
| 2 | 413 | 13.5 | 77.8 | 3.0 | 3.5 | 0.0 | 2.2 | 98.0 |
| 3 | 443 | 14.3 | 85.3 | 0.2 | 0.0 | 0.0 | 0.2 | 98.8 |
| 4 | 473 | 14.5 | 81.0 | 0.2 | 0.2 | 4.3 | 0.0 | 97.0 |

Anisole could not be converted when HZSM-5 was applied alone as a catalyst under 2 MPa of H2. However, when Pd/C and HZSM-5 were used jointly, the rate of C–O bond cleavage was enhanced steeply. Moreover, when only Pd/C was used as the catalyst, the selectivity of cyclohexane was only 0.6% and this value increased to 1.5% using the combined catalyst system (Table 3). These results suggest that the presence of dual catalytic function is favorable for the overall HDO. In addition, the acid sites of HZSM-5 are critical for breaking down the ether bond between the saturated carbon ring and the methyl group, as observed by increasing the amount of HZSM-5 from 0.1 g to 0.5 g (Table 3). The selectivity of cyclohexane increased drastically from 0.7 to 77.8%, accompanied by a decrease in methoxy-cyclohexane and cyclohexanol. When the amount of reactant was increased by a factor of six, hydrogenation was incomplete, leaving 44.1% of the anisole unconverted. A large amount of cyclohexanone was detected. The selectivity of cyclohexane as a function of temperature follows the same trend (Fig. 1). It was found that when the reaction temperature was below 413 K, the amount of cyclohexane present was under the detection limit. However, when the reaction temperature was further increased above 413 K, the selectivity of cyclohexane improved significantly, especially for hydrocarbons derived from anisole (79.4%). Moreover, as the temperature was further increased to 443 K, the HDO of phenol and anisole to cyclohexane proceeded quantitatively at high rates with intermediates barely detected.
| Entry | Amount of reactant and catalysts | Selectivity of products (C%) |
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Anisole (mol) | HZSM-5 (g) | Pd/C (g) | CH3OH |
|
|
|
|
|
||
| a Typical reaction conditions: the amount of anisole, HZSM-5 (Si/Al = 15) and Pd/C was added as displayed above, H2O (20 mL), 2 MPa H2, 413 K, 2 h, stirred at 680 rpm. | ||||||||||
| 1 | 0.005 | 0.50 | 0.05 | 13.5 | 77.8 | 3.0 | 3.5 | 0.0 | 2.2 | 98.0 |
| 2 | 0.005 | 0.10 | 0.05 | 9.1 | 0.7 | 36.0 | 53.7 | 0.2 | 0.3 | 99.3 |
| 3 | 0.03 | 0.10 | 0.05 | 8.3 | 1.1 | 42.0 | 24.4 | 24.2 | 0.0 | 55.9 |
| 4 | 0.03 | 0.02 | 0.10 | 5.6 | 0.2 | 61.1 | 18.8 | 14.2 | 0.0 | 65.8 |
| 5 | 0.03 | 0.02 | 0.20 | 4.3 | 0.5 | 69.7 | 20.0 | 5.5 | 0.0 | 96.6 |
| 6 | 0.03 | 0.00 | 0.20 | 5.3 | 0.3 | 62.9 | 20.3 | 11.1 | 0.0 | 87.1 |

 | ||
| Fig. 1 Selectivities of the main products over Pd/C and HZSM-5 as a function of reaction temperature using mono-substituted monomers as the reactants: (a) phenol, (b) anisole. Typical reaction conditions: reactant (0.005 mol), HZSM-5 (Si/Al = 15, 0.5 g), Pd/C (0.05 g), H2O (20 mL), 2 MPa H2, 2 h, stirred at 680 rpm. | ||
3.3 HDO of multi-substituted phenolics
Depending on the biomass source and the pyrolysis process conditions, phenolic compounds represent a significant fraction of the total bio-oil. Among them, methoxy phenols (guaiacol, syringol and their derivatives) are particularly abundant. Therefore, it is important to identify the reaction pathways that methoxy phenols may undergo on zeolites. According to previous considerations, guaiacol, which possesses two types of C–O bonds, namely Csp2–OH (hydroxyl) and Csp2–OCH3 (methoxy), was chosen as a model compound to probe the catalyst behavior with respect to these two organic functions. Similar to phenol conversion, the main products, 2-methoxy-cyclohexanol and 2-methoxy-cyclohexanone, were formed due to the fast hydrogenation of the aromatic ring. In all cases, the conversion of guaiacol was lower than that of phenol. 1,2-Cyclohexanediol and 2-hydroxyl-cyclohexanone derived from the hydrolysis of the methoxy group were not detected even at 443 K (Fig. 2a), pointing to the lower reactivity of guaiacol. This can be attributed to the steric constraint imposed when the active metal centre approaches the C–O bonds46 and the electron-donating character of the hydroxyl group that can stabilize the transition state carbocation and decrease the hydrolysis rate of the methoxy group. In addition, high temperatures may facilitate the hydrolysis of methoxy groups, leading to the formation of 1,2-cyclohexanediol and 2-hydroxyl-cyclohexanone. However, the conversion of these products originating from the hydrolysis of methoxy groups was hindered.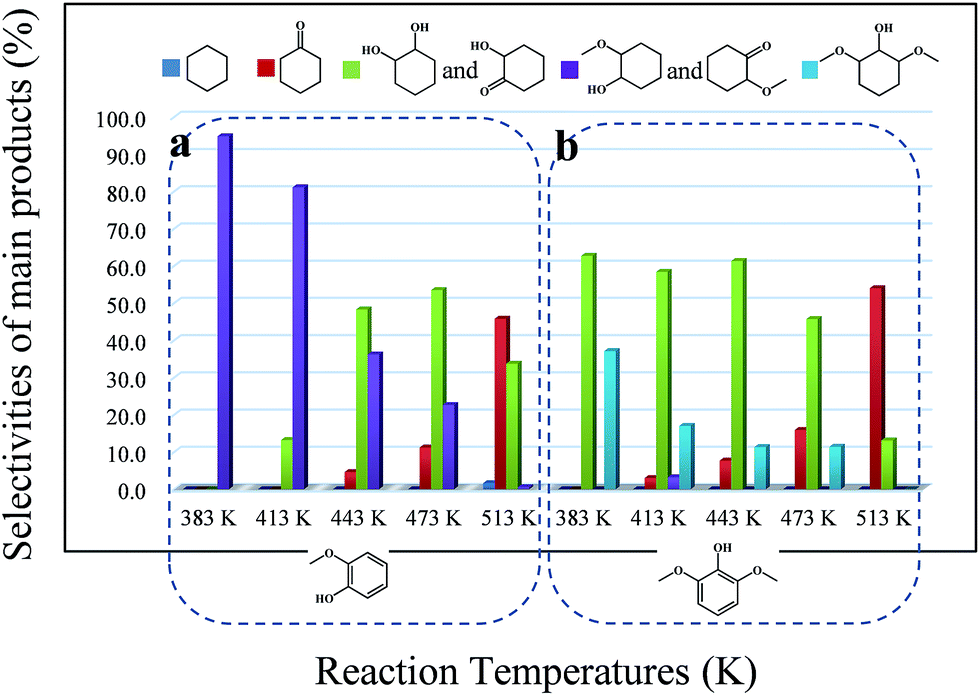 | ||
| Fig. 2 Selectivities of the main products over Pd/C and HZSM-5 as a function of reaction temperature using multi-substituted phenolic compounds as the reactants: (a) guaiacol, (b) 2,6-dimethoxy-phenol. Typical reaction conditions: reactant (0.005 mol), HZSM-5 (Si/Al = 15, 0.5 g), Pd/C (0.05 g), H2O (20 mL), 2 MPa H2, 2 h, stirred at 680 rpm. | ||
The selectivity of cyclohexane was only 5.9% at 513 K (Tables 4 and S2†), while the selectivity of the products containing one oxygen functional group reached 54.0%, with cyclohexanone being the largest percentage. The high selectivity for cyclohexanone indicates the higher adsorption constant of the aromatic rings, compared to that of the saturated cyclic compounds.
| Entry | Reaction temperature (K) | Selectivity of products (C%) |
|
|||
|---|---|---|---|---|---|---|
| CH3OH |
|
Products with one oxygen-containing functional group | Products with two oxygen-containing functional groups | |||
| a Typical reaction conditions: guaiacol (0.005 mol), HZSM-5 (Si/Al = 15, 0.5 g), Pd/C (0.05 g), H2O (20 mL), 2 MPa H2, 2 h, stirred at 680 rpm. | ||||||
| 1 | 383 | 0.9 | 0.0 | 5.8 | 93.2 | 39.4 |
| 2 | 413 | 3.0 | 0.0 | 7.9 | 89.1 | 100.0 |
| 3 | 443 | 9.6 | 0.0 | 10.4 | 80.1 | 86.4 |
| 4 | 473 | 11.5 | 0.0 | 18.3 | 70.1 | 83.0 |
| 5 | 513 | 14.3 | 5.9 | 54.0 | 25.7 | 77.4 |

Guaiacol was hydrodeoxygenated through the reaction pathway shown in Scheme 2. The aromatic ring of guaiacol was fully hydrogenated above 383 K and further deoxygenated to cyclohexanol, cyclohexanone and cyclohexane when heated to 513 K. Because hydrogenation of the aromatic ring of guaiacol was preferred at lower temperature, the formation of phenol was not observed, despite the fact that phenol could be formed via the demethylation and dehydration of guaiacol.7 Because of the excellent hydrogenation ability of Pd/C, the reaction pathway (guaiacol to 2-methoxycyclohexanol to hydrodeoxygenation products) proposed in Scheme 2 is similar to that reported by Lercher et al.43 and is quite different from some other reported catalytic systems (guaiacol to phenol to hydrodeoxygenation products).45–48
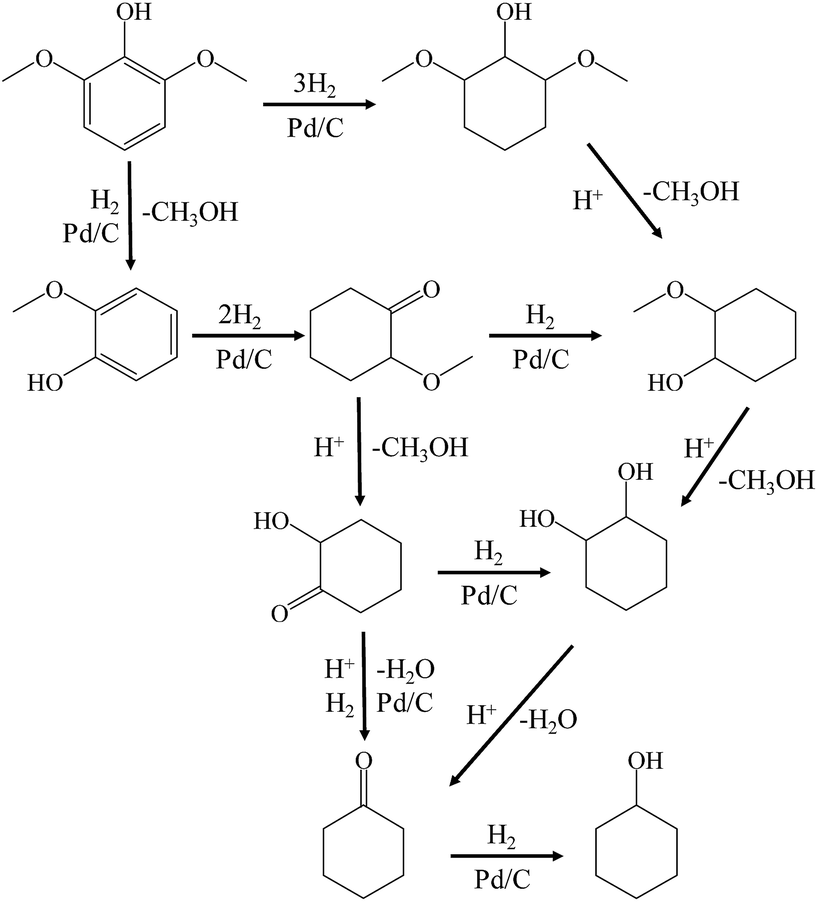 | ||
| Scheme 2 Reaction pathways for the conversion of 2,6-dimethoxy-phenol to cyclohexane over Pd/C and HZSM-5 in the liquid phase. | ||
As indicated in Table S3,† the HDO of 2,6-dimethoxy-phenol, with one hydroxyl group and two adjacent methoxy groups, is much more difficult. Cyclohexane was not observed even at 513 K (see the ESI†). In addition, the conversion of 2,6-dimethoxy-phenol was the lowest compared with phenol and guaiacol. The main products included 2-hydroxyl-cyclohexanone, 1,2-cyclohexanedione, 1,2-cyclohexanediol and 2,6-dimethoxy-cyclohexanol. From the product distributions, it is obvious that the selectivity of the products with one oxygen-containing functional group increased with temperature, showing the opposite trend to those of the products with two and three oxygen-containing functional groups (Table 5). The oxygen removal ratio of three phenolic model compounds (phenol, guaiacol and 2,6-dimethoxy-phenol) increased as the temperature increased (Fig. 3), which suggests that higher temperature is favorable for the removal of oxygen-containing functional groups. At 513 K, 2,6-dimethoxy-cyclohexanol disappeared, while 2-hydroxyl-cyclohexanone and cyclohexanone were the primary products, coinciding with those of guaiacol. The selectivity of the products with one oxygen-containing functional group reached 54.0% at 513 K.
| Entry | Reaction temperature (K) | Selectivity of products (C%) |
|
|||
|---|---|---|---|---|---|---|
| CH3OH | Products with one oxygen-containing functional group | Products with two oxygen-containing functional groups | Products with three oxygen-containing functional groups | |||
| a Typical reaction conditions: 2,6-dimethoxy-phenol (0.005 mol), HZSM-5 (Si/Al = 15, 0.5 g), Pd/C (0.05 g), H2O (20 mL), 2 MPa H2, 2 h, stirred at 680 rpm. | ||||||
| 1 | 383 | 18.8 | 0.0 | 57.1 | 24.0 | 6.5 |
| 2 | 413 | 21.9 | 5.6 | 62.6 | 10.0 | 89.2 |
| 3 | 443 | 23.2 | 9.6 | 61.0 | 6.2 | 53.1 |
| 4 | 473 | 23.0 | 19.4 | 51.1 | 6.4 | 66.9 |
| 5 | 513 | 24.4 | 65.9 | 9.6 | 0.0 | 73.1 |
| 6 | 413 K with Pd/C and beta (Si/Al = 12.5) | 19.0 | 3.9 | 70.4 | 6.9 | 92.3 |
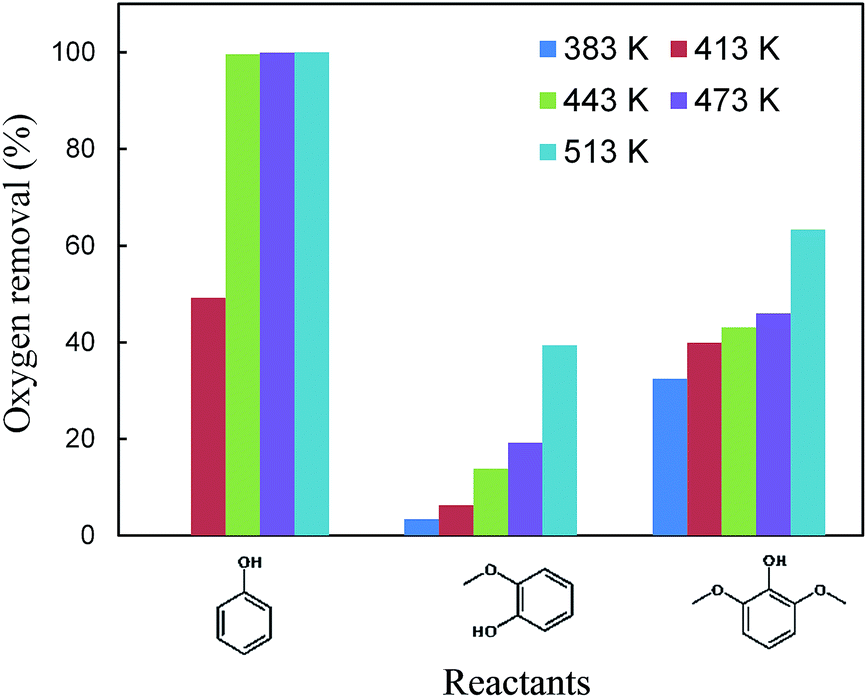 | ||
| Fig. 3 Oxygen removal over Pd/C and HZSM-5 as a function of reaction temperature using phenol, guaiacol and 2,6-dimethoxy-phenol. Typical reaction conditions: reactant (0.005 mol), HZSM-5 (Si/Al = 15, 0.5 g), Pd/C (0.05 g), H2O (20 mL), 2 MPa H2, 2 h, stirred at 680 rpm. | ||
3.4 The influence of 1,2-cyclohexanediol on the further HDO of multi-substituted phenolic compounds
A very large fraction of the hydrodeoxygenation products from guaiacol and 2,6-dimethoxy-phenol was dominated by 2-hydroxyl-cyclohexanone and 1,2-cyclohexanediol. The conversion of 2-hydroxyl-cyclohexanone to 1,2-cyclohexanediol with a double bond could be accomplished via ketoenol tautomerism. Therefore, the removal of the hydroxyl groups of cyclohexanediol must be the key reaction and we can hypothesize that this reaction was hindered by the presence of cyclohexanediol with or without a double bond. To validate this hypothesis, a group of comparative experiments were performed and the results are summarized in Tables 6 and 7.| Reaction temperature (K) | Selectivity of products (C%) |
|
|||||
|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
||
| a Typical reaction conditions: trans-1,2-cyclohexanediol (0.005 mol), HZSM-5 (Si/Al = 15, 0.5 g), Pd/C (0.05 g), H2O (20 mL), 2 MPa H2, 2 h, stirred at 680 rpm. | |||||||
| 413 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 100.0 | 1.3 |
| 513 | 2.3 | 0.0 | 93.6 | 0.0 | 4.1 | 0.0 | 100.0 |
| 513 K without Pd/C | 0.0 | 0.0 | 14.3 | 1.2 | 81.8 | 2.6 | 38.3 |
| 513 K without HZSM-5 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |

| Reaction temperature (K) | Selectivity of products (C%) |
|
|||
|---|---|---|---|---|---|
|
|
|
|
||
| a Typical reaction conditions: phenol (0.005 mol), trans-1,2-cyclohexanediol (0.0025 mol), HZSM-5 (Si/Al = 15, 0.5 g), Pd/C (0.05 g), H2O (20 mL), 2 MPa H2, 2 h, stirred at 680 rpm. | |||||
| 413 | 0.0 | 33.0 | 66.9 | 0.1 | 75.2 |
| 443 | 0.0 | 41.0 | 58.5 | 0.5 | 75.6 |
| 473 | 43.5 | 6.7 | 49.8 | 0.0 | 54.0 |
| 513 | 50.2 | 0 | 49.8 | 0.0 | 99.9 |
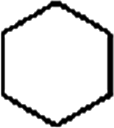
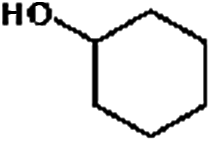
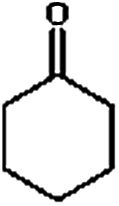
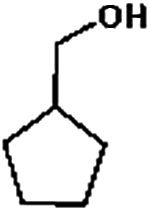
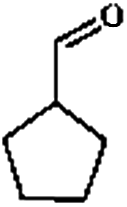
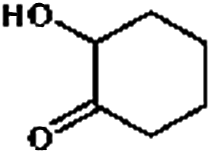
Firstly, the HDO of 1,2-cyclohexanediol was conducted over Pd/C and HZSM-5 (Table 6). At 413 K, no cyclohexane could be detected in the product. 1,2-Cyclohexanediol was completely converted to 2-hydroxyl-cyclohexanone, implying that it is rather difficult to cleave the C–O bonds of the saturated alcohol. With the temperature increased to 513 K, cyclohexane could be detected (about 2.3%), but the product was dominated by cyclohexanone (about 93.6%), suggesting that dehydration is favorable at higher temperature. This result is very similar to those obtained for guaiacol and 2,6-dimethoxy-phenol. It is most probable that the further hydrogenation of cyclohexanone during the conversion of guaiacol and 2,6-dimethoxy-phenol was inhibited. To test the respective functions of the combined catalyst, the HDO performance of Pd/C and HZSM-5 for 1,2-cyclohexanediol was tested separately. It was found that 1,2-cyclohexanediol could not be converted without HZSM-5 even at 513 K, but could be converted to cyclopentanecarbaldehyde with a selectivity of 81.8% without Pd/C, verifying that HZSM-5 mainly plays a dehydration role under the current reaction conditions and HZSM-5 contributes to the isomerization. A reaction pathway can be proposed in which the hydroxyl group of 1,2-cyclohexanediol is first protonated by the Brønsted acid sites and then undergoes dehydration, isomerization, deprotonation and hydrogenation to form cyclopentyl-methanol. This reaction pathway is similar to that of 1,2-cyclohexanediol on Ni/HZSM-5.46
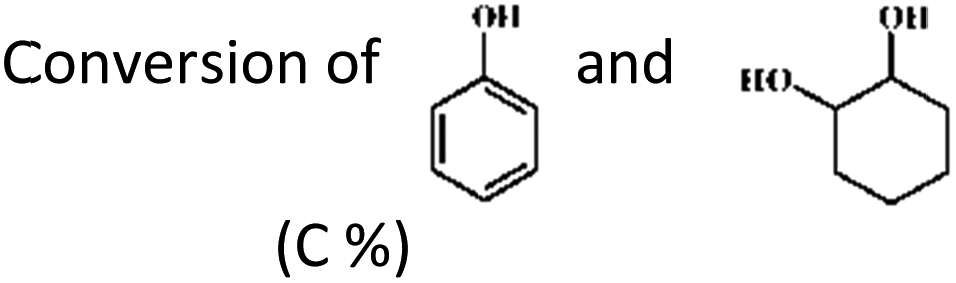
Secondly, the HDO of phenol and trans-1,2-cyclohexanediol (molar ratio: 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was conducted using the same reaction conditions (Table 7 and Fig. 4). As mentioned above, the catalyst system composed of Pd/C and HZSM-5 exhibited excellent reactivity when phenol was the sole reactant (Table 1). However, in the presence of 1,2-cyclohexanediol, almost no cyclohexane was detected at 413 K and 443 K (the selectivities of cyclohexane from phenol at 413 K and 443 K are 49.2 and 99.6%, respectively), confirming that the catalytic conversion of saturated cyclic alcohol was difficult and implying that the catalysts could be deactivated in the presence of 1,2-cyclohexanediol. According to the reaction mechanism (Scheme 1a), the benzene ring of phenol was first partially hydrogenated to form cyclohexanone, which could be further hydrogenated to form cyclohexanol and then dehydrated to cyclohexane. The hydrogenation of phenol to cyclohexanol was so fast that hardly any cyclohexanone as an intermediate was observed (Table 1). However, the selectivity of cyclohexanone, compared to the other products of phenol, was the highest when 1,2-cyclohexanediol was added into the reactant. Thus, the primary conclusion can be made that under the likely operation of this two-step mechanism, the Pd/C–ZSM-5 system was very active during the first step but not active during the second step under the experimental conditions. It is likely that the catalysts were deactivated by 1,2-cyclohexanediol through the formation of unreactive protonated species between 1,2-cyclohexanediol and the Brønsted acid sites of HZSM-5, similar to the previously reported dimers that were proposed to explain the dehydration of 2-butanol on a POM cluster.49 In other words, the strong hydrogen interactions inhibited the further hydrogenation of cyclohexanone. Additionally, the moderate hydrogen pressure employed in the present contribution also contributed to the domination of cyclohexanone, because low hydrogen pressure can stabilize the ketone intermediate and suppress cycloalkane formation.26
:1) was conducted using the same reaction conditions (Table 7 and Fig. 4). As mentioned above, the catalyst system composed of Pd/C and HZSM-5 exhibited excellent reactivity when phenol was the sole reactant (Table 1). However, in the presence of 1,2-cyclohexanediol, almost no cyclohexane was detected at 413 K and 443 K (the selectivities of cyclohexane from phenol at 413 K and 443 K are 49.2 and 99.6%, respectively), confirming that the catalytic conversion of saturated cyclic alcohol was difficult and implying that the catalysts could be deactivated in the presence of 1,2-cyclohexanediol. According to the reaction mechanism (Scheme 1a), the benzene ring of phenol was first partially hydrogenated to form cyclohexanone, which could be further hydrogenated to form cyclohexanol and then dehydrated to cyclohexane. The hydrogenation of phenol to cyclohexanol was so fast that hardly any cyclohexanone as an intermediate was observed (Table 1). However, the selectivity of cyclohexanone, compared to the other products of phenol, was the highest when 1,2-cyclohexanediol was added into the reactant. Thus, the primary conclusion can be made that under the likely operation of this two-step mechanism, the Pd/C–ZSM-5 system was very active during the first step but not active during the second step under the experimental conditions. It is likely that the catalysts were deactivated by 1,2-cyclohexanediol through the formation of unreactive protonated species between 1,2-cyclohexanediol and the Brønsted acid sites of HZSM-5, similar to the previously reported dimers that were proposed to explain the dehydration of 2-butanol on a POM cluster.49 In other words, the strong hydrogen interactions inhibited the further hydrogenation of cyclohexanone. Additionally, the moderate hydrogen pressure employed in the present contribution also contributed to the domination of cyclohexanone, because low hydrogen pressure can stabilize the ketone intermediate and suppress cycloalkane formation.26
 | ||
| Fig. 4 Conversion and selectivities of the main products over Pd/C and HZSM-5 as a function of reaction temperature using different reactants: (a) only phenol (0.005 mol) as the reactant, (b) 1,2-cyclohexanediol (0.0025 mol) added into phenol (0.005 mol). Typical reaction conditions: reactant (0.005 mol), HZSM-5 (Si/Al = 15, 0.5 g), Pd/C (0.05 g), H2O (20 mL), 2 MPa H2, 2 h, stirred at 680 rpm. | ||
4. Conclusions
In conclusion, the catalytic upgrading of lignin-derived phenolic monomers into hydrocarbons via HDO over a bi-functional catalyst system Pd/C–HZSM-5 has been studied. The possible reaction pathways and the respective roles of Pd/C and HZSM-5 were revealed. The HDO of phenol to cyclohexane proceeded via an initial metal-catalyzed aromatic ring hydrogenation, naphthenic alcohol dehydration, and subsequently metal-catalyzed cycloalkene hydrogenation. Anisole was also converted into methoxy-cyclohexane via hydrogenation. In the HDO of guaiacol, guaiacol was firstly hydrogenated to 2-methoxy-cyclohexanone and then converted to cyclohexane via sequential hydrolysis, dehydration and hydrogenation. For the HDO of 2,6-dimethoxy-phenol, the hydrogenation of the aromatic ring and the direct removal of the methoxy groups from the aromatic ring proceeded in parallel with 2-methoxy-cyclohexanol and guaiacol as the respective intermediates. The effects of temperature and substitution of the benzene ring on the HDO were also investigated systematically. For simple phenolic monomers like phenol and anisole, the HDO can be accomplished with very high selectivity to generate cyclohexane at elevated temperature. However, for multi-substituted monomers, only traces of hydrocarbons can be obtained, even above 513 K. The low conversion of guaiacol and 2,6-dimethoxy-phenol may be due to the steric constraint imposed when the active metal center approaches the C–O bonds, in addition to the inhibition of the electron-donating hydroxyl group that can stabilize the transition state carbocation. A large number of by-products was obtained, including cyclohexanone, 2-hydroxyl-cyclohexanone and 1,2-cyclohexanediol, in the HDO products of guaiacol and 2,6-dimethoxy-phenol. The comparative experiments performed on 1,2-cyclohexanediol with or without the presence of phenol clearly demonstrate that the HDO of guaiacol and 2,6-dimethoxy-phenol is inhibited by the presence of cyclohexanediol, which can form unreactive protonated species and inhibit the further HDO of guaiacol and 2,6-dimethoxy-phenol.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 21276263, 21306214, 21171173) and the “100 Talents” program of the Chinese Academy of Sciences (Grant No. KJCX2-EW-H05).References
- D. L. Klass, Chem. Eng. News, 2002, 22, 5 Search PubMed.
- D. L. Klass, Biomass for Renewable Energy, Fuels and Chemicals, Academic Press, San Diego, 1998, p. 30 Search PubMed.
- R. D. Perlack and L. L. Wright, Energy, 1995, 20, 279–284 CrossRef.
- K. S. Tyson, Fuel Cycle Evaluations of Biomass-Ethanol and Reformulated Gasoline; Report No. NREL/TP-263–2950, DE94000227, National Renewable Energy Laboratory, Golden, CO, 1993 Search PubMed.
- C. E. Wyman, Appl. Biochem. Biotechnol., 1994, 45/46, 897–915 CrossRef.
- L. R. Lynd, J. H. Cushman, R. J. Nichols and C. E. Wyman, Science, 1991, 251, 1318–1323 CAS.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- Q. Zhang, J. Chang, T. Wang and Y. Xu, Energy Convers. Manage., 2007, 48, 87–92 CrossRef CAS.
- A. Brems, R. Dewil, J. Baeyens, J. P. K. Seville, C. Prayogo and G. Bending, The future of biomass pyrolysis in the production of value-added products, in Proceedings of the SDEWES Conference, Dubrovnik, September 2011, pp. 26–29 Search PubMed.
- G. Burcu, K. Osman, H. J. H. Erik, A. P. Evgeny and J. M. H. Emiel, Catal. Today, 2014, 233, 83–91 CrossRef.
- R. Rinaldi and F. Schuth, Energy Environ. Sci., 2009, 2, 610–628 CAS.
- S. De, B. Saha and R. Luque, Bioresour. Technol., 2015, 178, 108–118 CrossRef CAS PubMed.
- Z. He and X. Wang, Catal. Sustainable Energy Prod., 2012, 1, 28 Search PubMed.
- B. Quan, L. Hanwu, H. Z. Alan, W. Lu, R. Shoujie, L. Jing, W. Yi, L. Yupeng, T. Juming, Z. Qin and R. Roger, Bioresour. Technol., 2012, 124, 470–477 CrossRef PubMed.
- P. Teerawit, S. Manish, M. Karthick and R. L. Yuriy, Energy Environ. Sci., 2014, 7, 2660–2669 Search PubMed.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 CAS.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 CAS.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, R. Aguado and J. Bilbao, Ind. Eng. Chem. Res., 2004, 43, 2610–2618 CrossRef CAS.
- P. D. Chantal, S. Kaliaguine and J. L. Grandmaison, Appl. Catal., 1985, 18, 133–145 CrossRef CAS.
- P. A. Horne and P. T. Williams, Renewable Energy, 1996, 7, 131–144 CrossRef CAS.
- M. A. Peralta, T. Sooknoi, T. Danuthai and D. E. Resasco, J. Mol. Catal. A: Chem., 2009, 312, 78–86 CrossRef CAS.
- S. Jin, Z. Xiao, X. Chen, L. Wang, J. Guo, M. Zhang and C. Liang, Ind. Eng. Chem. Res., 2015, 54, 2302–2310 CrossRef CAS.
- J. D. Adjaye and N. N. Bakhshi, Biomass Bioenergy, 1995, 8, 131–149 CrossRef CAS.
- C. Zhao, J. He, A. A. Lemonidou, X. Li and J. A. Lercher, J. Catal., 2011, 280, 8–16 CrossRef CAS.
- J. He, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 20768–20775 CrossRef CAS PubMed.
- C. Zhao and J. A. Lercher, ChemCatChem, 2012, 4, 64–68 CrossRef CAS.
- C. Zhang, J. Xing, L. Song, H. Xin, S. Lin, L. Xing and X. Li, Catal. Today, 2014, 234, 145–152 CrossRef CAS.
- Q. Song, J. Cai, J. Zhang, W. Yu, F. Wang and J. Xu, Chin. J. Catal., 2013, 34, 651–658 CrossRef CAS.
- C. R. Lee, J. S. Yoon, Y. W. Sun, J. W. Choi, J. M. Ha, D. J. Suh and Y. K. Park, Catal. Commun., 2012, 17, 54–58 CrossRef CAS.
- S. Jin, Z. Xiao, C. Li, X. Chen, L. Wang, J. Xing, W. Li and C. Liang, Catal. Today, 2014, 234, 125–132 CrossRef CAS.
- H. Ren, C. Li, D. Yin, J. Liu and C. Liang, RSC Adv., 2016, 6, 85659–85665 RSC.
- X. Wang and R. Rinaldi, ChemSusChem, 2012, 5, 1455–1466 CrossRef CAS PubMed.
- C. Zhao and J. A. Lercher, Angew. Chem., Int. Ed., 2012, 51, 5935–5940 CrossRef CAS PubMed.
- J. Horacek, G. St'avova, V. Kelbichova and D. Kubicka, Catal. Today, 2013, 204, 38–45 CrossRef CAS.
- J. D. Adjaye and N. N. Bakhshi, Fuel Process. Technol., 1995, 45, 185–202 CrossRef CAS.
- R. K. Sharma and N. N. Bakhshi, Can. J. Chem. Eng., 1991, 69, 1071–1081 CrossRef CAS.
- C. A. Chen, H. Pakdel and C. Roy, Bioresour. Technol., 2011, 79, 277–299 CrossRef.
- M. Kleinert and T. Barth, Chem. Eng. Technol., 2008, 31, 736–745 CrossRef CAS.
- P. D. Wild, R. V. Laan, A. Kloekhorst and H. J. Heeres, Environ. Prog. Sustainable Energy, 2009, 28, 461–469 CrossRef.
- Y. Nakagawa, M. Ishikawa, M. Tamura and K. Tomishige, Green Chem., 2014, 16, 2197–2203 RSC.
- W. Schutyser, G. Van den Bossche, A. Raaffels, S. Van den Bosch, S.-F. Koelewijn, T. Renders and B. F. Sels, ACS Sustainable Chem. Eng., 2016, 4, 5336–5346 CrossRef CAS.
- M. J. Girgis and B. C. Gates, Ind. Eng. Chem. Res., 1991, 30, 2021–2058 CrossRef CAS.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Angew. Chem., 2009, 48, 3987–3990 CrossRef CAS PubMed.
- X. Zhu, R. G. Mallinson and D. E. Resasco, Appl. Catal., A, 2010, 379, 172–181 CrossRef CAS.
- Y.-C. Lin, C.-L. Li, H.-P. Wan, H.-T. Lee and C.-F. Liu, Energy Fuels, 2011, 25, 890–896 CrossRef CAS.
- W. Song, Y. Liu, E. Baráth, C. Zhao and J. A. Lercher, Green Chem., 2015, 17, 1204–1218 RSC.
- R. N. Olcese, M. Bettahar, D. Petitjean, B. Malaman, F. Giovanella and A. Dufour, Appl. Catal., B, 2012, 115–116, 63–73 CrossRef CAS.
- M. V. Bykova, D. Y. Ermakov, V. V. Kaichev, O. A. Bulavchenko, A. A. Saraev, M. Y. Lebedev and V. A. Yakovlev, Appl. Catal., B, 2012, 113–114, 296–307 CrossRef CAS.
- J. Macht, M. J. Janik, M. Neurock and E. Iglesia, Angew. Chem., Int. Ed., 2007, 46, 7864–7868 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra22492j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |