
Isatin pentafluorophenylhydrazones: interesting conformational change during anion sensing†
M. Horvátha,
M. Cigáň*a,
J. Filoa,
K. Jakusováa,
M. Gáplovskýb,
R. Šándrika and
A. Gáplovskýa
aFaculty of Natural Sciences, Institute of Chemistry, Comenius University, Mlynská dolina CH-2, Ilkovičova 6, SK-842 15 Bratislava, Slovakia. E-mail: horvathm@fns.uniba.sk; cigan@fns.uniba.sk; filo@fns.uniba.sk; jakusova@fns.uniba.sk; sandrik@fns.uniba.sk; gaplovsky@fns.uniba.sk
bDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Comenius University, Odbojárov 10, SK-832 32 Bratislava, Slovakia. E-mail: gaplovsky@fpharm.uniba.sk
First published on 11th November 2016
Abstract
The anion sensing properties of two new easily synthesized isatin pentafluorophenylhydrazone reversible colorimetric chemosensors are studied herein. The F− or CH3COO− anion addition to isatin pentafluorophenylhydrazone solutions in aprotic organic solvents results in hydrazone NH group deprotonation of the initial Z-hydrazo form (acid–base keto/enolate reaction), with significant equilibrium shift to the more conjugated E-azo enolate side. Initial solutions thus turn orange. This fast equilibrium is followed by slow conformational change to the more stable E-azo enolate conformer with the configuration of the E-hydrazo isomer. Interestingly, the F− or CH3COO− detection in semi-aqueous media leads directly to slow formation of the second E-azo enolate conformer, without the observation of an initial fast equilibrium between Z-hydrazo and the corresponding first E-azo enolate conformer. Although the reaction time in semi-aqueous media thus increases to several minutes (tens of minutes), the advantage of isatin pentafluorophenylhydrazone chemosensors still remains their easy synthesis and their reversibility. The determined sensitivity towards F− and CH3COO− anions is among the highest published sensitivity to these anions in organic solvents and allows confident F− detection at the tolerated drinking-water fluoride level in semi-aqueous media.
Introduction
Ion and neutral molecule recognition and signalling is currently one of the most intense emerging areas of supramolecular chemistry. While anions play a very important role in many chemical and biological processes, their identification and quantification is often complicated; especially in biological systems. Since one way to solve this problem is to utilize new optical sensors (receptors) specifically designed for individual anions, the focus of supramolecular chemistry over the last twenty years has centred on the development of selective colorimetric or fluorescent anion sensors.1–5 Research in this area continues today in the work of Wenzel, Chudzinski, Bergamaschi, Jiménez, Martínez-Máñez, Gale and others.6–23Anions interact with sensors particularly through hydrogen bonding interactions.4,13,24–26 Due to the facile synthesis and easily tunable NH acidity, amides, ureas and thioureas are the most widely employed hydrogen bond donor groups in anion sensor systems, and this has not changed during last three years.11,12 Detection of anions in aqueous environments is currently the most interesting target in the chemosensor field.12 This is, however, unachievable for most designs relying in hydrogen bonding, since even minute amounts of water disrupt these interactions. Moreover, sensor molecules are often complex constructs and several synthetic steps are required for their preparation.11,27 The last trend in the chemosensor field is the common cation and anion detection using one sensor molecule (dual sensors).28,29
Anion sensing, tautomerism and the light and thermally initiated mutual E- and Z-isomer transformations of eleven efficient and easily synthesized isatin phenylsemicarbazone colorimetric sensors Ia-XIa were investigated in our few recent studies.30–35 Addition of strongly basic F− and CH3COO− anions led to the most acidic NH group deprotonation and the Ia-XIa E-isomers transformed from their self-associate keto-form to monomer enolate form. Sensors provided an excellent signal to noise ratio and the utilization of both isomers significantly enlarges the detection range. Detection of F− or CH3COO− anions at high weakly-basic anion excess was also possible. Moreover, the low photochemical E–Z isomerization efficiency allowed reliable detection of strongly basic anions. Detection limits of 3–4 × 10−7 mol dm−3 for F− and CH3COO− anions are among the lowest published detection limits for these anions in organic solvents. Furthermore, due to excellent E-isomer sensitivity in organic media, these isomers can be used for F− or CH3COO− sensing in semi-aqueous media and allow confident F− detection at tolerated fluoride drinking-water level.
Herein, we focused on research of isatin pentafluorophenylhydrazones (Scheme 1). In the last three years, hydrazones found important application in various areas of supramolecular chemistry as molecular switches, photo- and thermo-sensitive supramolecular arrangement and colorimetric or fluorescent chemosensors.36–40 Our recent two studies investigated the photochromic properties of basic isatin phenylhydrazones and isatin diphenylhydrazones as a new type of molecular switches.41,42 Isatin presence in the structure shifts their absorption to the Vis-region of the electromagnetic spectrum, and the isomerization around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond bestows them a simple on–off photochromic character. Contrary to the corresponding isatin diphenylhydrazones, the low switching amplitude in absorbance in isatin phenylhydrazones (with approximately 20–30% conversion in the photostationary state) hamper their photochromic applications.42 However, the addition of strongly basic anion to phenylhydrazone solutions led to hydrazine NH group deprotonation and created a new diazene T-type Vis photochromic system with sufficiently separated absorption maxima of the corresponding two forms. Although interesting photochromic behaviour with the thermally stable E-azo form and a quite unstable Z-azo form could lead to several interesting photochromic applications, the hydrazine NH group deprotonation sensitivity was insufficient to produce effective F− or CH3COO− colorimetric sensors.
N double bond bestows them a simple on–off photochromic character. Contrary to the corresponding isatin diphenylhydrazones, the low switching amplitude in absorbance in isatin phenylhydrazones (with approximately 20–30% conversion in the photostationary state) hamper their photochromic applications.42 However, the addition of strongly basic anion to phenylhydrazone solutions led to hydrazine NH group deprotonation and created a new diazene T-type Vis photochromic system with sufficiently separated absorption maxima of the corresponding two forms. Although interesting photochromic behaviour with the thermally stable E-azo form and a quite unstable Z-azo form could lead to several interesting photochromic applications, the hydrazine NH group deprotonation sensitivity was insufficient to produce effective F− or CH3COO− colorimetric sensors.
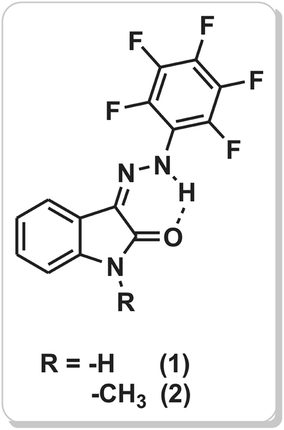 | ||
| Scheme 1 Molecular structure of studied isatin pentafluorophenylhydrazones 1 and 2. | ||
In this research, the anion sensing properties and interesting conformational changes of two easily synthesized isatin pentafluorophenylhydrazones 1 and 2 are reported. Due to the more significant difference between absorption maxima of corresponding protonated and deprotonated forms and more acidic character of hydrazone (aniline) NH hydrogen, isatin pentafluorophenylhydrazones 1 a 2 are approximately 10-times more sensitive to strongly basic anions compared to basic isatin phenylhydrazones and can compete in sensitivity with isatin phenylsemicarbazones. Compared to these chemosensors, the advantage of isatin pentafluorophenylhydrazones is the insensitivity of their selectivity to solution dilution.
Results and discussion
Spectral characteristics
Similar to basic isatin phenylhydrazones, only more stable Z-hydrazo isomers with strong intramolecular hydrogen bonding were isolated from the reaction mixture during synthesis (Table 1 and Fig. 1).| Isomer | ΔG kJ mol−1 |
|---|---|
| Z-Hydrazo | 0 |
| E-Hydrazo | 32 |
| E-Azo enol | 41 |
| Z-Hydrazo/E-azo enol TS | 37 |
| Z-Azo-enol | 127 |
| E-Azo-enol conformer | 53 |
| Z-π-Hydrazo | 38 |
| Z-Hydrazo/Z-π-hydrazo TS | 76 |
 | ||
| Fig. 1 19F–1H HOESY spectrum of 2 in DMSO-d6 (T = 298.15 K) and schematic structure suggesting through space H–F interaction in more stable Z-hydrazo tautomer (Z-isomer). | ||
Due to opposite electron-withdrawing effect of isatin and pentafluorophenyl structural subunits, isatin pentafluorophenylhydrazone chromophores 1 and 2 exhibit hypsochromically shifted absorption bands (maxima) compared to their unsubstituted phenyl analogues (ESI Fig. S1†). Interestingly, presence of long-wavelength (LW) absorption band shoulder is evident in absorption spectrum of methyl derivative 2, contrary to basic methylated isatin phenylhydrazone (ESI Fig. S1†). Its modest indication can be observed also in absorption spectrum of non-methylated pentafluorophenylhydrazone 1. The shoulder/band intensity ratio increases further with a gradual temperature increase to a constant value at a given temperature (Fig. 2).
 | ||
| Fig. 2 UV-Vis spectrum of isatin pentafluorophenylhydrazone 1 in DMF at various temperatures and its reversible photochemical transformation (c = 1 × 10−4 mol dm−3). | ||
The observed behaviour indicates keto/enol (hydrazo/azo) tautomeric equilibrium (Scheme 2). Increase in temperature shifts the hydrazo/azo tautomeric equilibrium to more conjugated E-azo form side, therefore temperature decrease should lead to back thermal reaction and thus to LW shoulder disappearance. However, solution cooling to room temperature results in an unexpected additional (although only modest) LW shoulder increase and clearly indicates the presence of at least one additional equilibrium. Due to the low overall Z-hydrazo form conversion (∼5%), this more complex behaviour will be discussed at the end of the next section. But it should be mentioned here that all developed forms of pentafluorophenylhydrazones 1 and 2 in solution heating/cooling cycle can be photochemically transformed back to initial Z-hydrazo forms (Fig. 2). Both Z-hydrazo and E-azo tautomers are non-fluorescent in nature.
 | ||
| Scheme 2 Temperature and light influence on hydrazo/azo (keto/enol) tautomeric equilibrium of 1 and 2. | ||
Contrary to basic isatin phenylhydrazones, photochemical transformation of pentafluorophenylhydrazone Z-hydrazo isomers to corresponding hypsochromically shifted E-hydrazo isomers is quite less effective (∼5–10 times; ESI Fig. S2†), with approximately only 3–5% conversion in the photostationary state (in all used solvents: DMF, DMSO, CHCl3, MeOH; λirr = 370 or 405 nm). Composition of the photostationary state depends on the ratio of quantum yields for the forward and backward isomerization reactions and that of the absorption coefficients. Because Z- and E-hydrazo isomers of isatin phenylhydrazones have similar absorption spectra and also extinction coefficients (Fig. 3 and ref. 31), the forward and backward isomerization reactions should significantly differ in their quantum yields (ΦZ→E and ΦE→Z, respectively).
 | ||
| Fig. 3 Calculated UV-Vis absorption spectra of isatin pentafluorophenylhydrazone 1 Z-isomer and corresponding isomeric and tautomeric forms (Scheme 2 and ESI Scheme S1;† M062x 6-31+g(dp) level with included solvent effect of DMF). | ||
We assume that markedly lower ΦZ→E results from efficient excited state intramolecular proton transfer (ESIPT) due to the presence of the six-membered intramolecular hydrogen bond. Electron-deficient pentafluorophenyl aromatic ring in pentafluorophenylhydrazones 1 and 2 reduces electron density on the hydrazone (aniline) nitrogen and thus increase acidity of the hydrazone NH hydrogen compared to basic isatin phenylhydrazones (Table S1 and ESI Fig. S3; please, see also ESI† pKa constant determinations). However, flash spectroscopic techniques should be used to finally determine a dominant de-excitation pathway(s) of Z-hydrazo forms.
Anion addition
The F− or CH3COO− anion addition to 1 or 2 DMF solution results in hydrazone NH group deprotonation (acid–base keto/enolate reaction), with significant equilibrium shift to the more conjugated E-azo enolate side (Fig. 4 and S4†). | ||
| Fig. 4 1H NMR spectrum of isatin pentafluorophenylhydrazone 2 in DMSO-d6 before and after TBA+F− addition (T = 298.15 K). | ||
The acid–base keto (hydrazo)/enolate (azo) equilibrium is accompanied by the decrease of the initial LW absorption band intensity and simultaneous appearance of the new bathochromically shifted absorption band at 450 nm (Fig. 5 and ESI Fig. S5 and S6†). Initial solution thus turns orange.
 | ||
| Fig. 5 UV-Vis spectrum of isatin pentafluorophenylhydrazones 1 and 2 after F− and CH3COO− addition in DMF (cSENSOR = 1 × 10−4 M). | ||
Contrary to relatively slow response of basic isatin phenylhydrazones (equilibrium is reached in 2 minutes), pentafluorophenyl chemosensors 1 and 2 exhibit practically immediate deprotonation followed by slow thermal process which leads to small hypsochromic (blue) shift of the rapidly formed E-azo enolate absorption band at 460 nm (without solution colour change) and to simultaneous additional absorption increase in the 400–500 nm spectral region (Fig. 6). Interestingly, both 1H and 19F NMR spectra indicate total conversion of the initial E-azo enolate to newly formed chemical species during the slow second process (Fig. 7, 8 and ESI Fig. S7–S9†). Irradiation of the final solution (with equilibrium composition after the slow second process) decreases overall spectrum intensity, but the incurred spectrum does not overlap with the spectrum of E-azo enolate solution at the end of the fast initial process (after F− addition – Fig. S10†). As shown in Fig. S11,† the photochemical process is thermally reversible and isatin pentafluorophenylhydrazones therefore exhibit T-type Vis photochromic behaviour that can be regulated by the anion amount (Fig. 6).
 | ||
| Fig. 6 UV-Vis spectrum of isatin pentafluorophenylhydrazone 1 after F− anion addition in DMF (cSENSOR = 5 × 10−5 M; (A) cF− = 5 × 10−5 M; (B) cF− = 1 × 10−2 M; M = mol dm−3; T = 303.15 K). | ||
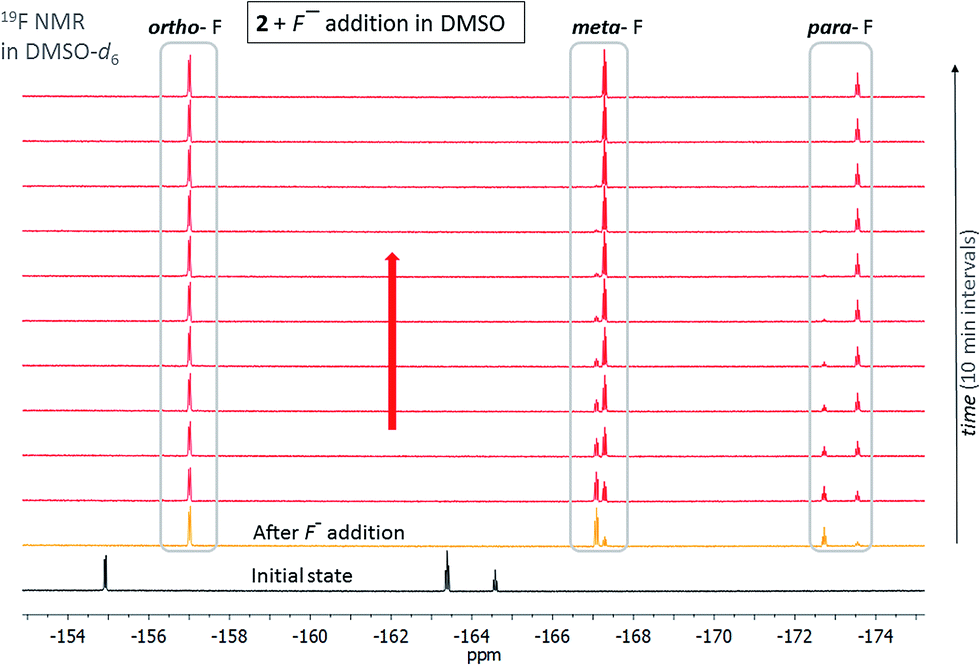 | ||
| Fig. 7 Evolution of the 19F NMR spectrum of isatin pentafluorophenylhydrazone 2 in DMSO-d6 after TBA+F− addition (T = 298.15 K). | ||
 | ||
| Fig. 8 Evolution of the 1H NMR spectrum of isatin pentafluorophenylhydrazone 2 in DMSO-d6 after TBA+F− addition (T = 298.15 K). | ||
Although the slow thermal process may suggest E/Z isomerization of the E-azo enolate to packed Z-azo enolate form (Scheme S1†), very low probable complete spontaneous E-azo enolate conversion to the less stable Z-azo enolate (Table 2) and low extinction coefficient of this new Z-azo form (Fig. 9) exclude thermal E/Z isomerization as the observed slow reaction pathway (Z-azo enolate is stabilized by lp-π interaction between the isatin oxygen lone pair and the pentafluorophenyl ring; with calculated O…centroid distance of 3.063 Å in vacuum).
| Isomer | ΔG kJ mol−1 |
|---|---|
| E-Azo-enolate | 17 |
| E-Azo-enolate conformer | 0 |
| E-Azo-enolate/E-azo-enolate conformer TS | 131 |
| Z-Azo-enolate | 42 |
| Z-Azo-enolate conformer | 55 |
| E-Azo enolate/Z-azo-enolate TS | 96 |
 | ||
| Fig. 9 Calculated UV-Vis absorption spectra of isatin pentafluorophenylhydrazone 1 Z-isomer and corresponding E- and Z-azo enolate forms (ESI Scheme S1;† M062x 6-31+g(dp) level with included solvent effect of DMF). | ||
Fortunately, 1H NMR spectra during the second process in both DMF and DMSO showed one interesting feature: increased doublet signal between 8.2–8.7 ppm at the expense of doublet at 7.2–7.6 ppm (Fig. 8 and ESI Fig. S9†). This doublet signal is characteristic for E-hydrazo form of isatin phenylhydrazones and belongs to C(4)-H hydrogens on E-hydrazo isomer isatin ring (please, see 1H NMR spectra of Z1 and Z2 hydrazo isomers after irradiation with Vis light in ref. 42). Quite significant shift of isatin C(4)–H doublet to lower fields reflects the difference in conjugation between Z-isomers 1 and 2 and the corresponding E-isomers. Although the E-hydrazo isomer cannot explain the previously discussed slow thermal process (Fig. 6), the characteristic doublet points out to structural similarity of the final and the E-hydrazo forms. Indeed, observed slow process can be rationalized by conformational change of the initial E-azo enolate to more stable E-azo enolate conformer (Scheme 3 and ESI Scheme S2†), similar to possible conformational equilibrium between two mono- (or differently) ortho-substituted azobenzene conformers.43
 | ||
| Scheme 3 Proposed F− anion sensing mechanism in aprotic organic solvents: deprotonation of Z-isomers 1 and 2 by strongly basic anion (acid–base keto/enolate equilibrium) followed by E-azo enolate form conformation change to more stable conformer. | ||
This second conformer has the same configuration as the E-hydrazo isomer. As shown in Fig. 3 and 9, the quantum-chemical calculations clearly suggest equal (or higher) extinction coefficient of the newly formed E-azo enolate (E-azo enol) conformer and its slightly hypsochromically shifted absorption maximum. Furthermore, calculated Gibbs free energies confirm higher stability of the second E-azo enolate conformer (Table 2). High transition state (TS) energy related to the E-azo-enolate transformation to the second conformer calculated in vacuum reflects the rotational motion of molecule with quite significant conjugation breaking during the conformational change (Table 2). Based on experimentally observed conformational rate and overall conversion, we assume that both DMSO and DMF as polar solvents additionally stabilized the corresponding TS and also the newly formed E-azo enolate conformer.
It should be noted here that weakly basic HSO4−, H2PO4−, Cl− and Br− anions practically do not affect the UV-Vis spectra of 1 and 2 solutions (ESI Fig. S12 and S13†).
Because the characteristic doublet signal above 8.0 ppm of the second E-azo enolate conformer was observed also in 1H NMR spectra of basic isatin phenylhydrazones (unfortunately, we omitted him in our previous paper), we can now generalize that F− anion addition to isatin phenylhydrazone solution in polar aprotic solvents leads to hydrazone NH group deprotonation followed by conformational change to more stable azo enolate with “E-hydrazo like” conformation.
Stability of the second azo conformer and high back conformational activation barrier can explain also the specific behaviour of studied isatin pentafluorophenylhydrazone solutions at heating/cooling cycle discussed in the previous chapter (Fig. 2). The temperature increase results in the E-azo enol form formation which slowly transforms to second more stable conformer (which is structurally close to E-hydrazo form – ESI Scheme S3†). Simultaneously, both E-azo enol conformers isomerize to corresponding significantly less stable Z-azo enol forms and further increase in temperature therefore results only in slight intensity increase of the bathochromic shoulder. Due to the high back activation barrier, the newly formed second E-azo enol conformer with high extinction does not transform back to the initial less stable E-azo enol conformer after cooling, whereas the less stable Z-azo enol form of the second conformer isomerizes during the cooling process back to the corresponding E-azo enol second conformer. Therefore, the final UV-Vis spectrum of cold solution after heating/cooling cycle contains more intense bathochromic shoulder than the heated or the initial solution (Fig. 2). Indeed, the presence of the characteristic E-hydrazo doublet and significant shift of both NH protons in the final 1H NMR spectrum of 1 at the end of heating/cooling cycle (compared to initial Z-hydrazo isomer 1H NMR spectrum) suggests the presence of the second E-azo enol conformer (Fig. S14†).
As already mentioned, all present equilibrium forms at the end of heating/cooling cycle can be photochemically transformed back to the initial Z-hydrazo tautomer (Fig. 2). In addition to previously discussed hydrazo and azo forms, equilibrium composition of the heated/cooled solution may complicate other stable chemical species like E-hydrazo and Z-π-hydrazo form (please, see Table S1 and ESI Scheme S3†).
Anion sensing
Due to significant difference between absorption maxima of keto and enolate forms and more acidic character of hydrazone (aniline) NH hydrogen (Table S1† – pKa values), isatin pentafluorophenylhydrazones 1 a 2 are more sensitive to presence of strongly basic anions compared to basic isatin phenylhydrazones (Table S2 – Kass values; ESI Fig. S15; please see also ESI† Association constant determinations).Because the conformational change of E-azo enolate forms in DMF is significantly slower compared to initial acid–base keto/enolate equilibrium, compounds 1 and 2 can be used as colorimetric sensors for strongly basic F− and CH3COO− anions. The calculated detection (3σ/S) and quantification (10σ/S) limits for F− and CH3COO− anions using the studied isatin pentafluorophenylhydrazone Z-isomer sensors are summarized in Table 3.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O solvent mixture (φr = 9:1) at 298.16 K (refer to absorbance at 450 nm; σ = 0.005)
:H2O solvent mixture (φr = 9:1) at 298.16 K (refer to absorbance at 450 nm; σ = 0.005)
| DMF | ||||
|---|---|---|---|---|
| Compd | 3σ/S F− (mol dm−3) | 10σ/S F− (mol dm−3) | 3σ/S CH3COO− (mol dm−3) | 10σ/S CH3COO− (mol dm−3) |
| a Detection (3σ/S) and quantification (10σ/S) limits for F− and CH3COO− anions in an original water sample are 10-times higher. | ||||
| 1 | 2.5 × 10−6 | 8.2 × 10−6 | 4.2 × 10−6 | 1.4 × 10−5 |
| 2 | 6.8 × 10−7 | 2.3 × 10−6 | 7.1 × 10−7 | 2.4 × 10−6 |
The determined 3σ/S for F− and CH3COO− anions by sensors 1 and 2 are amongst the lowest published detection limits for these anions in organic solvent (using the same σ value for 3σ/S determination)44 and are similar to previously published isatin phenylsemicarbazones.30 However, the advantage of isatin pentafluorophenylhydrazones is the insensitivity of their selectivity to solution dilution. Lower detection limit of methylated derivative 2 results from better E-azo enolate stabilization (Table S1† – pKa values).
As already mentioned, detection of anions in aqueous environments is currently the most interesting target in the chemosensor field.12 Studied isatin pentafluorophenylhydrazone chemosensors can also be used for F− or CH3COO− sensing in semi-aqueous media (Fig. 10).
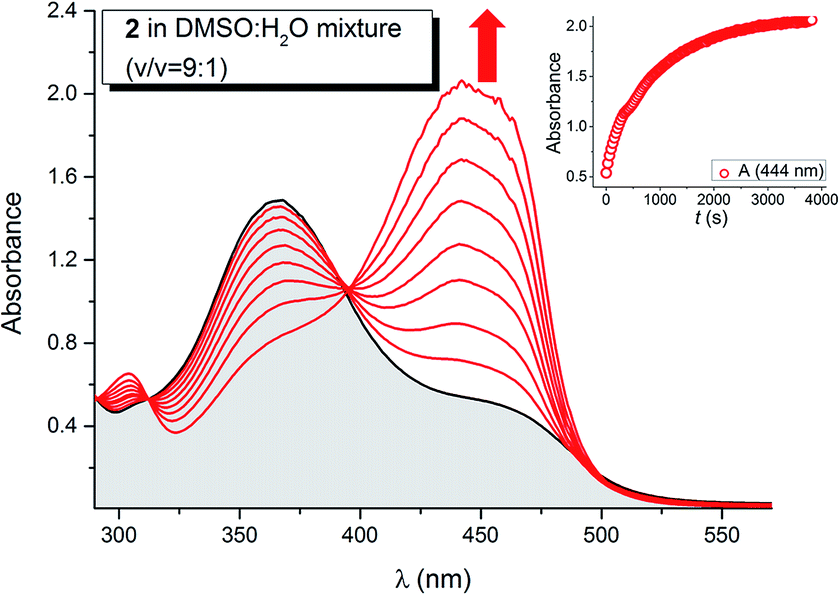 | ||
| Fig. 10 Evolution of the UV-Vis spectrum of isatin pentafluorophenylhydrazone 2 after F− anion addition in DMSO:H2O (v/v = 9:1) solvent mixture (cSENSOR = 1 × 10−4 M; cF− = 1 × 10−3 M; T = 293.15 K). Inset: corresponding evolution of absorbance at 450 nm. | ||
Interestingly, although addition of water markedly returns the present acid–base equilibrium to the Z-hydrazo form (ESI Fig. S16†), the F− or CH3COO− addition to 1 and 2 solution in DMF:H2O mixture (v/v = 9:1) leads directly to slow formation of the second E-azo enolate conformer, without the observation of initial fast equilibrium between Z-hydrazo and corresponding first E-azo enolate form. The evolution of 1H and 19F NMR spectra after F− addition to 1 and 2 solution in DMF:H2O (or DMSO:H2O) solvent mixture (ESI Fig. S17–S21†) and the comparison of 1H NMR spectra in pure DMF or DMSO with those in corresponding solvent mixture (ESI Fig. S22–S25†) clearly confirm this conclusion. Based on these facts we assume that the solvation by water stabilizes Z-hydrazo anionic form of isatin pentafluorophenylhydrazones after their deprotonation by TBA+F− and the stable Z-hydrazo anionic form then slowly directly transforms to the more stable second E-azo enolate conformer (Scheme 4).
 | ||
| Scheme 4 Proposed F− anion sensing mechanism in semi-aqueous media: deprotonation of Z-isomers 1 and 2 by strongly basic anion (acid–base equilibrium) in DMF:H2O or DMSO:H2O (v/v = 9:1) solvent mixture followed by slow thermal transformation of water-stabilized Z-hydrazo anionic form to the most stable E-azo enolate conformer. | ||
Therefore, after water sample addition to chemosensor DMF solution, the resultant solution mixture should stay few minutes before a quantitative analysis of F− or CH3COO− anions in the original water sample (ESI Fig. S26†). More reliable response and higher sensitivity can be obtained by monitoring of initial absorbance growth rate and by reaction time increase, respectively (ESI Fig. S26D†). Determined low detection limit for F− anion in 9:1 DMF:H2O mixture by both sensors allows confident F− detection at tolerated drinking-water fluoride level (1.5 mg L−1 to 0.17 mmol dm−3; Table 2). Moreover, the choice of an appropriate aprotic organic solvent also influences the response rate and the sensitivity of studied chemosensors (ESI Fig. S27†). Because the sensing mechanism has acid–base character, care must be taken when interpreting data in semi-aqueous media due to the CO2(g)/HCO3−/CO32− equilibrium (i.e. initial water sample should be degassed). Fig. 11 illustrates the qualitative detection of F− in real water sample. Unfortunately, studied commercial mouth-wash contains undefined amount of hydrochloric acid which suppresses isatin pentafluorophenylhydrazone acid–base response to fluoride anion. Therefore, the quantitative comparison to calibration curve in DMSO:H2O solvent mixture was not possible.
 | ||
| Fig. 11 Evolution of the UV-Vis spectrum of isatin pentafluorophenylhydrazone 2 in DMSO after commercial mouth-wash addition (cSENSOR = 1 × 10−4 M; cF− = 250 ppm = 4 × 10−3 M Na+F−; T = 293.15 K; M = mol dm−3; 1st spectrum was measured immediately after mouth wash addition). Inset: corresponding evolution of absorbance at 450 nm. | ||
Conclusions
This paper investigated the anion sensing properties of two new easily synthesized isatin pentafluorophenylhydrazone reversible colorimetric chemosensors based on acid–base keto (hydrazo)/enolate (azo) equilibrium. Due to their micromolar to sub-millimolar sensitivity, studied chemosensors can be useful in the detection of environmentally or biologically important F− and CH3COO− anions in both organic and semi-aqueous media. However, because of direct but slower production of the second more stable E-azo enolate conformer, the analysis of water samples in semi-aqueous media requests several minutes, contrary to practically immediate analysis in organic media. Moreover, care must be taken when interpreting data in semi-aqueous media due to the acid–base character of sensing mechanism. Although isatin pentafluorophenylhydrazone exhibit comparable detection limits with previously published isatin semicarbazones, the advantage of isatin pentafluorophenylhydrazones is the insensitivity of their selectivity to solution dilution (because sensing mechanism of isatin semicarbazones includes aggregate formation).Experimental section
Synthesis
Isatin N2-pentafluorophenylhydrazone 1 and isatin 1-methyl-N2-pentafluorophenylhydrazone 2 were prepared using a procedure by Vine et al.45 (Scheme 5). All chemicals was commercially available by Sigma Aldrich (isatin, 1-methylisatin, pentafluorophenylhydrazine, ethanol). Synthesis of unmethylated isatin N2-pentafluorophenylhydrazone 1 as potential anticonvulsant and antineoplastic agent was already published in ref. 46 and 47, however its anion sensing properties have not been investigated till now. Isatin 1-methyl-N2-pentafluorophenylhydrazone 2 is a new compound. For further data on the synthesis and characterization of isatin N2-pentafluorophenylhydrazone 1 and isatin 1-methyl-N2-pentafluorophenylhydrazone 2, please see ESI† Synthesis.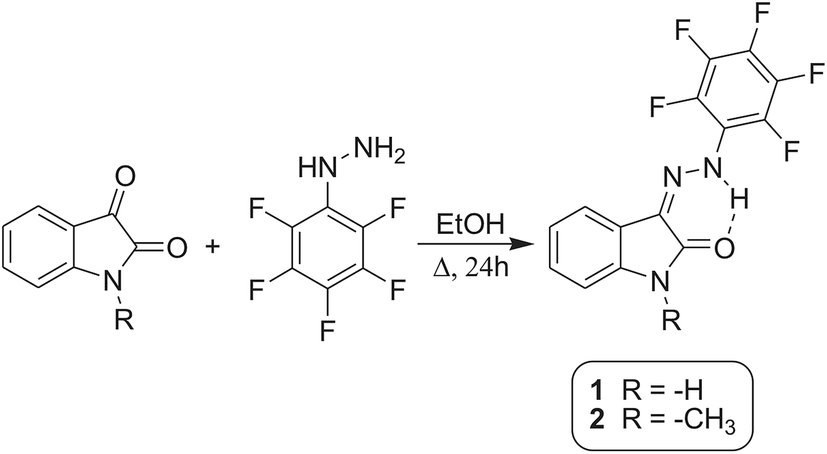 | ||
| Scheme 5 Synthesis of 1 and 2 Z-isomers in EtOH. | ||
Spectroscopic measurements
Electronic absorption spectra were obtained on a HP 8452A diode array spectrophotometer (Hewlett Packard, USA). DMF as a solvent (Acros Organics, 99.5%, For Analysis, Belgium) was used without further purification. Solution absorbance was measured in a 1 cm quartz cuvette.Titration experiments
:distilled H2O solvent mixture (φr = 9:1) at 298.16 K. The isatin pentafluorophenylhydrazone solutions were titrated with the corresponding anion solutions to obtain 1 × 10−4 mol dm−3 overall sensor concentrations of the resultant solution. The titration process in the anion concentration range of 1 × 10−5 mol dm−3 to 1 × 10−2 mol dm−3 was monitored by UV-Vis spectroscopy (in a 1 cm cuvette).
 | (1) |
 | (2) |
:H2O solvent mixture (φr = 9:1), sensor absorbance A (as a function of anion concentration) was measured at 15th minute after A− anion addition (ESI Fig. Y1†).Acknowledgements
This work was supported by the Slovak Grant Agency for Science (VEGA grant no. 1/0463/15).Notes and references
- P. J. Smith, M. V. Reddington and C. S. Wilcox, Tetrahedron Lett., 1992, 33, 6085–6088, DOI:10.1016/S0040-4039(00)60012-6.
- E. Fan, S. A. van Arman, S. Kincaid and A. D. Hamilton, J. Am. Chem. Soc., 1993, 115, 369–370, DOI:10.1021/ja00054a066.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76, DOI:10.1002/1521-3773(20020104)41.
- M. Boiocchi, L. D. Boca, D. E. Gómez, L. Fabbrizzi, L. Licchelli and E. Monzani, J. Am. Chem. Soc., 2004, 126, 16507–16514, DOI:10.1021/ja045936c.
- S. E. García-Garrido, C. Caltagirone, M. E. Light and P. A. Gale, Chem. Commun., 2007, 1450–1452, 10.1039/b618072h.
- M. Wenzel, M. E. Light, A. P. Davis and P. A. Gale, Chem. Commun., 2011, 47, 7641–7643, 10.1039/c1cc12439k.
- M. G. Chudzinski, C. A. Corey, A. McClary and M. S. Taylor, J. Am. Chem. Soc., 2011, 133, 10559–10567, DOI:10.1021/ja202096f.
- G. Bergamaschi, M. Boiocchi, E. Monzani and V. Amendola, Org. Biomol. Chem., 2011, 9, 8276–8283, 10.1039/c1ob06193c.
- M. B. Jiménez, V. Alcázar, R. Paláez, F. Sanz, A. L. Fuentes de Arriba and M. C. Caballero, Org. Biomol. Chem., 2012, 10, 1181–1185, 10.1039/c1ob06540h.
- L. E. Santos-Figueroa, M. E. Moragues, E. Climent, A. Agostini, R. Martínez-Máñez and F. Sancenón, Chem. Soc. Rev., 2013, 42, 3489–3613, 10.1039/c3cs35429f.
- P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205–241, 10.1039/c3cs60316d.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516, DOI:10.1002/1521-3773(20010202)40.
- B. Garg, T. Bisht and S. M. S. Chauhan, Sens. Actuators, B, 2012, 168, 318–328, DOI:10.1016/j.snb.2012.04.029.
- A. Rostami and M. S. Taylor, Macromol. Rapid Commun., 2012, 33, 21–34, DOI:10.1002/marc.201100528.
- K. Kaur, R. Saini, A. Kumar, V. Luxami, N. Kaur, P. Singh and S. Kumar, Coord. Chem. Rev., 2012, 256, 1992–2028, DOI:10.1016/j.ccr.2012.04.013.
- Y. Zhou, J. F. Zhang and J. Yoon, Chem. Rev., 2014, 114, 5511–5571, DOI:10.1021/cr400352m.
- N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716–11754, DOI:10.1002/anie.201309937.
- P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2015, 44, 4212–4227, 10.1039/c4cs00179f.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 5511–5571, DOI:10.1021/acs.chemrev.5b00099.
- F. Sancenûn, L. Pascual, M. Oroval, E. Aznar and R. Martínez-Máñez, ChemistryOpen, 2015, 4, 418–437, DOI:10.1002/open.201500053.
- Y. Jiao, B. Zhu, J. Chen and X. Duan, Theranostics, 2015, 5, 173–187, DOI:10.7150/thno.9860.
- P. A. Gale, E. N. W. Howe and X. Wu, Chem., 2016, 1, 351–422, DOI:10.1016/j.chempr.2016.08.004.
- S. K. Sahoo, G.-D. Kim and H.-J. Choi, J. Photochem. Photobiol., C, 2016, 27, 30–53, DOI:10.1016/j.jphotochemrev.2016.04.004.
- S. L. A. Kumar, M. S. Kumar, P. B. Sreeja and A. Sreekanth, Spectrochim. Acta, Part A, 2013, 113, 123–129, DOI:10.1016/j.saa.2013.04.103.
- S. O. Kang, R. A. Begum and K. Bowman-James, Angew. Chem., Int. Ed., 2006, 45, 7882–7894, DOI:10.1002/anie.200602006.
- S. Hu, Y. Guo, J. Xu and S. Shao, Spectrochim. Acta, Part A, 2009, 72, 1043–1046, DOI:10.1016/j.saa.2008.12.042.
- C. B. Rosen, D. J. Hansen and K. V. Gothelf, Org. Biomol. Chem., 2013, 11, 7916–7922, 10.1039/c3ob41078a.
- C. Kar, S. Samanta, S. Mukherjee, B. K. Datta, A. Ramesh and G. Das, New J. Chem., 2014, 38, 2660–2669, 10.1039/c4nj00239c.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981, 10.1039/c3cs60385g.
- M. Cigáň, K. Jakusová, J. Donovalová, V. Szöcs and A. Gáplovský, RSC Adv., 2014, 4, 54072–54079, 10.1039/c4ra04847d.
- K. Jakusová, M. Cigáň, J. Donovalová, M. Gáplovský, R. Sokolík and A. Gáplovský, J. Photochem. Photobiol., A, 2014, 288, 60–69, DOI:10.1016/j.jphotochem.2014.05.004.
- K. Jakusová, J. Donovalová, M. Cigáň, M. Gaplovský, V. Garaj and A. Gaplovský, Spectrochim. Acta, Part A, 2014, 123, 421–429, DOI:10.1016/j.saa.2013.12.073.
- K. Jakusová, J. Donovalová, M. Gaplovský, M. Cigáň, H. Stankovičová and A. Gaplovský, J. Phys. Org. Chem., 2013, 26, 805–813, DOI:10.1002/poc.3164.
- M. N. Chaur, D. Collado and J.-M. Lehn, Chem.–Eur. J, 2011, 17, 248–258, DOI:10.1002/chem.201002308.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981, 10.1039/c3cs60385g.
- G. Vantomme and J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 3940–3943, DOI:10.1002/anie.201210334.
- G. Vantomme and J.-M. Lehn, Chem.–Eur. J., 2014, 20, 1–7, DOI:10.1002/chem.201404561.
- G. Vantomme, S. Jiang and J.-M. Lehn, J. Am. Chem. Soc., 2014, 136, 9509–9518, DOI:10.1021/ja504813r.
- L. A. Tatum, X. Su and I. Aprahamian, Acc. Chem. Res., 2014, 47, 2141–2149, DOI:10.1021/ar500111f.
- Y. Yang, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 13190–13193, DOI:10.1021/ja508125n.
- M. Cigáň, K. Jakusová, J. Donovalová, V. Szöcs and A. Gáplovský, RSC Adv., 2015, 5, 62449–62459, 10.1039/c5ra06625e.
- M. Cigáň, K. Jakusová, M. Gáplovský, J. Filo, J. Donovalová and A. Gáplovský, Photochem. Photobiol. Sci., 2015, 14, 2064–2073, 10.1039/c5pp00275c.
- J. García-Amorós and D. Velasco, Beilstein J. Org. Chem., 2012, 8, 1003–10017, DOI:10.3762/bjoc.8.113.
- N. R. Chereddy, P. Nagaraju, M. V. N. Raju, K. Saranraj, S. Thennarasu and V. J. Rao, Dyes Pigm., 2015, 112, 201–209, DOI:10.1016/j.dyepig.2014.07.004.
- K. L. Vine, J. M. Locke, M. Ranson, K. Benkendorff, S. G. Pyne and J. B. Bremner, Bioorg. Med. Chem., 2007, 15, 931–938, DOI:10.1016/j.bmc.2006.10.035.
- F. D. Popp, J. Med. Chem., 1969, 12, 182–184, DOI:10.1021/jm00301a054.
- F. D. Popp, J. Heterocycl. Chem., 1984, 21, 1641–1645, DOI:10.1002/jhet.5570210614.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra22396f |
| This journal is © The Royal Society of Chemistry 2016 |