
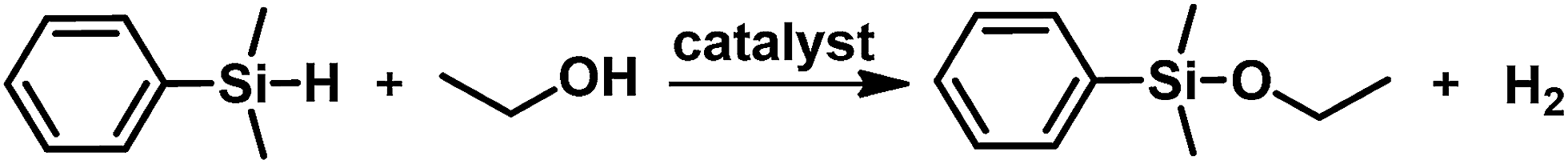
Silica-supported ultra small gold nanoparticles as nanoreactors for the etherification of silanes†
Cui Wang,
Xijie Lin,
Yuzhen Ge,
Zameer Hussain Shah,
Rongwen Lu* and
Shufen Zhang
State Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian, People's Republic of China. E-mail: lurw@dlut.edu.cn
First published on 11th October 2016
Abstract
Ultra small gold nanoparticles supported by porous silica (Au–SiO2) were successfully synthesized. Due to enrichment of reactants by silica, the Au–SiO2 particles functioned as nanoreactors for catalytic etherification of silanes with high selectivity and reusability. The reaction kinetics indicated that the catalysis operated by a zero order reaction mechanism, which is contrary to previously reported homogeneous catalysts, as well as Au–Al2O3 and Au–FeOx prepared by the same method. The mechanism of the reaction was described by the Langmuir–Hinshelwood model, with the rate determining step being the surface reaction on the gold nanoparticles.
Introduction
Supported metal nanoparticles have been the subject of immense interest.1,2 A specific metal nanoparticle can demonstrate distinctly different properties depending on the support employed.3,4 Porous silica is a widely used support for nanomaterials; significant contributions to various applications have been made using such materials, particularly in the field of heterogeneous catalysis.5–7 Organic synthesis is a key discipline under the umbrella of heterogeneous catalysis8 and etherification of silanes is a very important organic chemical reaction.9 Metal nanoparticles supported on silica may serve as highly efficient catalysts for this reaction.Etherification of silanes yields silyl ethers, which are important reagents and protecting groups for alcohols in organic synthesis.10,11 One strategy for achieving etherification of silanes is by employing a suitable catalyst.12–14 Among various available catalysts, Au-based nano catalysts have shown promising results.2 Raffa and coworkers15 reported the first catalytic application of Al2O3-supported Au nanoparticles in silane etherification at a temperature of 100 °C. In a subsequent study, surface functionalized Au nanoparticles with self-assembled monolayers of alkane thiols were found to be highly efficient catalysts for the same reaction at room temperature.16 The alkane thiol monolayers facilitated interaction between Au nanoparticles and the substrate molecules by creating a space for their encapsulation. This type of reactivity may be described as “nanoreactor”17 catalysis, in which a unique nano-scaled reaction space is partitioned from the bulk chemical environment. However, recycling of an organically stabilized catalyst is a daunting challenge.1 Au nanoparticles confined in a solid space with nano-scale dimensions, i.e., inside “nanoreactors”, offer better catalytic performance and allow for facile recovery and recycling. Silica is one of the most commonly used porous solids, and it has attracted a great deal of attention owing to its wide range of applications as adsorption material18–20 and catalytic support for metal nanoparticles.21–23 Silica also offers a high affinity for silanes,24 which makes it a very promising support candidate for their etherification. It is expected that gold supported on silica would exhibit a high performance like nanoreactors for the catalytic etherification of silanes.
Herein, we describe the synthesis of ultra small gold nanoparticles supported on porous silica (Au–SiO2) and evaluate their catalytic performance for the etherification of silanes at mild temperature. Our results showed that Au–SiO2 was a highly efficient catalyst. The effect of FeOx and Al2O3 supports on the catalytic activity of the Au nanoparticles was also evaluated. It was found that the choice of support had a profound effect on the catalytic activity. Au–SiO2 was found to work as a nanoreactor, providing superior catalysis due to better adsorption of the substrate molecules onto silica. We hope our findings will trigger an exploration of the role of different supports in the other chemical reactions that are catalyzed by supported metal nanoparticles.
Experimental
Materials
Polyoxyethylene (20) cetyl ether (Brij®58), methyldiphenylsilane (Ph2MeSiH), and triethylsilane (Et3SiH) were purchased from J&K. Chloroauric acid, tetraethyl orthosilicate (TEOS), ammonium hydroxide (NH3·H2O, 25–28%), sodium borohydride (NaBH4), cyclohexane, n-heptane, methanol, ethanol, isopropanol (IPA), n-butanol and benzyl alcohol were all purchased from Sinopharm Chemical Reagent Co., Ltd. (SCRC). All chemicals were analytical grade.Synthesis of Au–SiO2
The synthesis of Au–SiO2 was carried out in a reverse microemulsion system that consisted of 3.37 g Brij®58 and 15 mL of cyclohexane at 52 °C. Aqueous chloroauric acid (0.1 M, 0.2 mL) was added into the system with stirring. After 5 min, a NaBH4 solution (0.14 M, 0.7 mL) was added dropwise; the reaction conditions were then maintained for 30 min. Finally, 0.5 mL NH3·H2O and 1.0 mL TEOS were introduced to initiate the sol–gel process to prepare the silica support. The Au–SiO2 sample was collected by centrifugation at 6000 rpm for 10 min, and washed 3 times with IPA. The product was dried at 100 °C for 10 h and calcined at 500 °C in an air flow for 2 h.For the aggregation of small gold nanoparticles, the sample was calcined at 600 °C for 3 h under H2/N2 (5% H2, 95% N2) with a flow rate of 80 mL min−1.
For the synthesis of Au nanoparticles with the size of 4.0 nm, the experiment conditions were similar to the ultra small Au–SiO2; the only difference was the concentration and volume of added NaBH4 solution (0.1 M, 0.3 mL). Au nanoparticles with a size of 4.0 nm were denoted as Au (4.0 nm)–SiO2.
The role of the support was investigated using Al2O3 and FeOx as alterative supports for the Au nanoparticles. The synthesis followed the same steps as described for ultra small Au–SiO2. Aluminum isopropoxide and iron butyrate were used as precursors for aluminum and iron supports, respectively.
Etherification of silanes
The preparation of ethoxydimethylphenylsilane was used as a representative example. Tetrahydrofuran (THF, 6 mL), ethanol (0.2 mL), dimethylphenylsilane (0.1 mL) and nanocatalyst (20 mg, 0.2 mol% Au) were added to a reactor at room temperature. An Agilent 7000B Triple Quadrupole GC-MS instrument with a HP-5MS capillary column (30 m × 0.25 mm × 0.25 μm) and MS detector was used for the analysis, with He as carrier gas. The temperature program for the detection of ethoxydimethylphenylsilane was set from 60 °C (maintained for 2 min) to 180 °C, with a 15 °C min temperature ramp. The reaction progress was monitored by GC with a HP-5 capillary column using N2 as carrier gas and an FID detector, and the temperature program was the same as for GC-MS.For other silyl ethers, different temperatures were required, the details of which can be found in Table 2. The products were also detected by GC-MS with the only difference being the ceiling temperature.
|
||||
|---|---|---|---|---|
| Catalyst | Conversion (%) | Selectivity (%) | TOF (h−1) | |
| a Reaction conditions: silane (0.1 mL), alcohol (0.2 mL), THF (6.0 mL), catalyst (0.2 mol% Au), at room temperature for 2 hours. The conversion was obtained by GC-MS with internal standard, and TOF was calculated based on the whole gold atoms. | ||||
| 1 | Ultra small Au–SiO2 | >99 | >99 | 234 |
| 2 | Au–SiO2 (4.0 nm) | 56.0 | >99 | 126 |
| 3 | Au–Al2O3 (3.8 nm) | 22.2 | >99 | 55.5 |
| 4 | Au–FeOx (4.0 nm) | 7.5 | >99 | 18.8 |
|
|||||
|---|---|---|---|---|---|
| Silane | Alcohol | Conditions | Conversion (%) | Selectivity (%) | TOF (h−1) |
| a Reaction conditions: silane (0.1 mL), alcohol (0.2 mL), THF (6.0 mL), Au–SiO2 (20 mg). The reactions were performed under air and the conversion was obtained by GC-MS with internal standard. TOF was calculated based on the whole gold atoms. | |||||
| PhMe2SiH | Methanol | 25 °C, 50 min | >99 | >99 | 568 |
| Ph2MeSiH | Methanol | 25 °C, 1 h | >99 | >99 | 366 |
| Et3SiH | Methanol | 25 °C, 1.75 h | >99 | >99 | 260 |
| PhMe2SiH | Ethanol | 25 °C, 2 h | >99 | >99 | 234 |
| Ph2MeSiH | Ethanol | 25 °C, 2.5 h | >99 | >99 | 146 |
| Et3SiH | Ethanol | 50 °C, 7 h | 96.5 | >99 | 63 |
| PhMe2SiH | n-Butanol | 50 °C, 5 h | >99 | >99 | 94 |
| Ph2MeSiH | n-Butanol | 50 °C, 5 h | >99 | >99 | 73 |
| Et3SiH | Benzyl alcohol | 80 °C, 9 h | 91.7 | >99 | 46 |
| Ph2MeSiH | Benzyl alcohol | 80 °C, 9 h | 5 | >99 | — |

Characterization
Transmission electron microscopy (TEM) was performed by JEOL JEM-2000 EX using an accelerating voltage of 120 kV. UV-vis absorbance spectroscopy was carried out using an Agilent 8453. X-ray diffraction (XRD) patterns were recorded on a Rigaku DMAX IIIVC with Cu Kα (0.1542 nm) radiation, scanning from 10° to 80° (2 theta) at the rate of 8° min−1. X-ray photoelectron spectroscopy (XPS) was performed using a Thermo Fisher Scientific Eacalab 250Xi, and all the binding energies were calibrated with C 1s peak at 284.6 eV. The Au content was determined using an inductively coupled plasma-optical emission spectrometer, Perkin Elmer Optima 2000 (ICP). The surface area measurement was done using a Quantachrome Autosorb-1 MP surface area and pore size analyzer. The etherification progress was detected using an Agilent 7000B Triple Quadrupol gas chromatography tandem mass spectrometry (GC-MS) instrument.Results and discussion
Ultra small Au–SiO2 catalyst was prepared according to the sol–gel procedure in a reverse microemulsion system. The gold loading was determined to be 1.37 wt% by inductively coupled plasma spectrometry (ICP). Morphological analysis was carried out by TEM. As shown in Fig. 1a, the silica nanospheres were mono-dispersed with an average diameter of 30 ± 1.6 nm. The dark black points dispersed over silica were identified as ultra small Au nanoparticles of a size less than 1 nm. The surface plasmon resonance (SPR) peak of gold nanoparticles in the UV-vis absorbance spectrum (Fig. 2) was very weak, which confirmed their small size.25 It was also difficult to find diffraction peaks of Au in the XRD pattern due to the ultra small size of the particles.26 Therefore, calcination of Au–SiO2 under hydrogen was done to aggregate the gold particles (Fig. S1†). In comparison to the weak SPR peak of ultra small Au–SiO2, the aggregated Au–SiO2 showed a stronger absorption peak around 530 nm. These results proved the existence of the ultra small Au nanoparticles in the original sample. Fig. 1c shows the XRD pattern of the aggregated Au–SiO2. The expected diffraction peaks appeared and their positions matched well with the standard diffraction pattern of Au (JCPDS: 04-0784). The hump between 15° and 35° could be attributed to amorphous silica. This also indirectly proved the existence of Au and SiO2 in the original sample.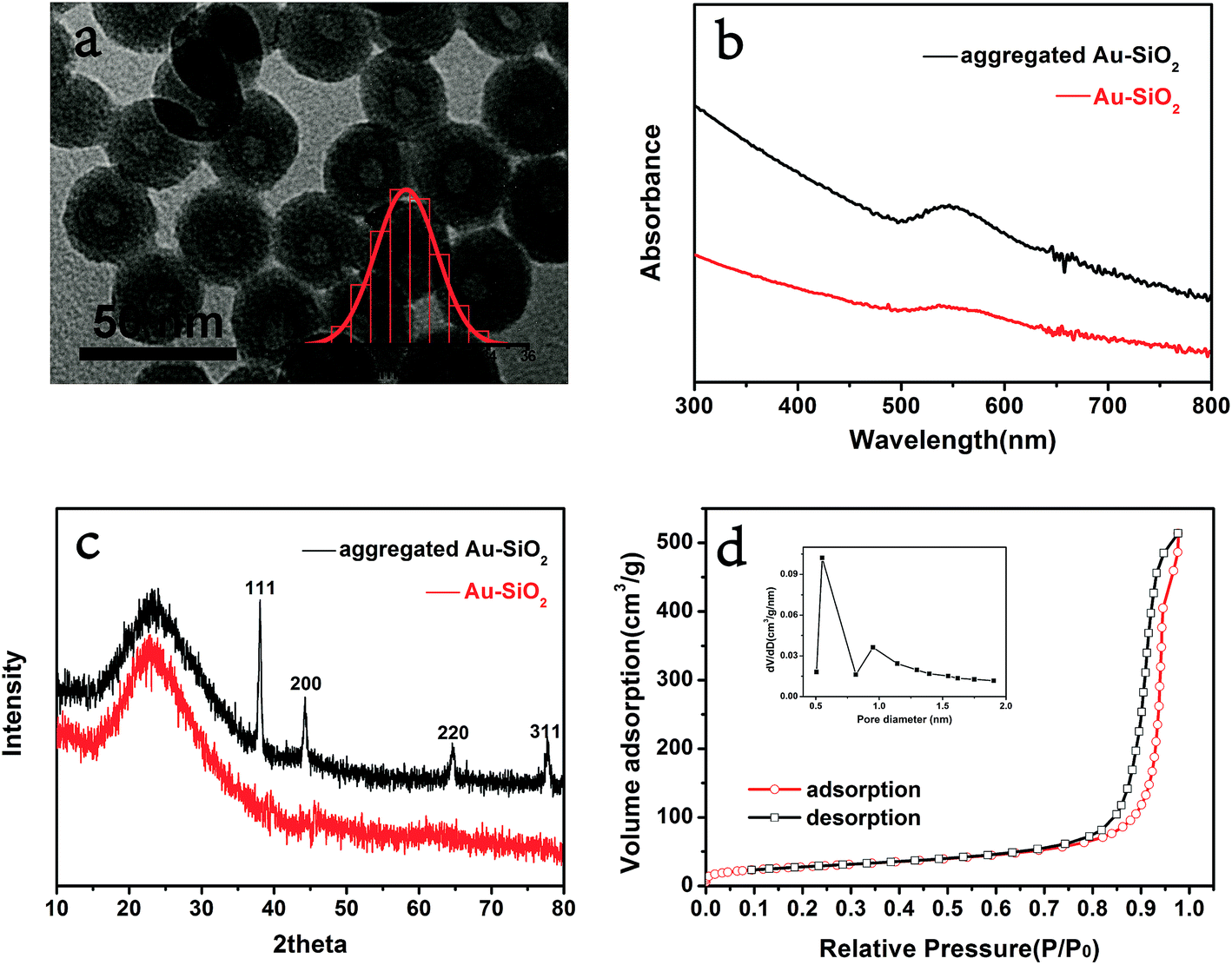 | ||
| Fig. 1 (a) TEM image of Au–SiO2 (inset showing the size distribution of silica); (b) UV-vis spectrum of the water solution of ultra small Au–SiO2 and aggregated Au–SiO2; (c) XRD pattern of the Au–SiO2 and aggregated Au–SiO2; (d) N2 sorption isotherm of Au–SiO2 (inset showing the distribution of micropore size). | ||
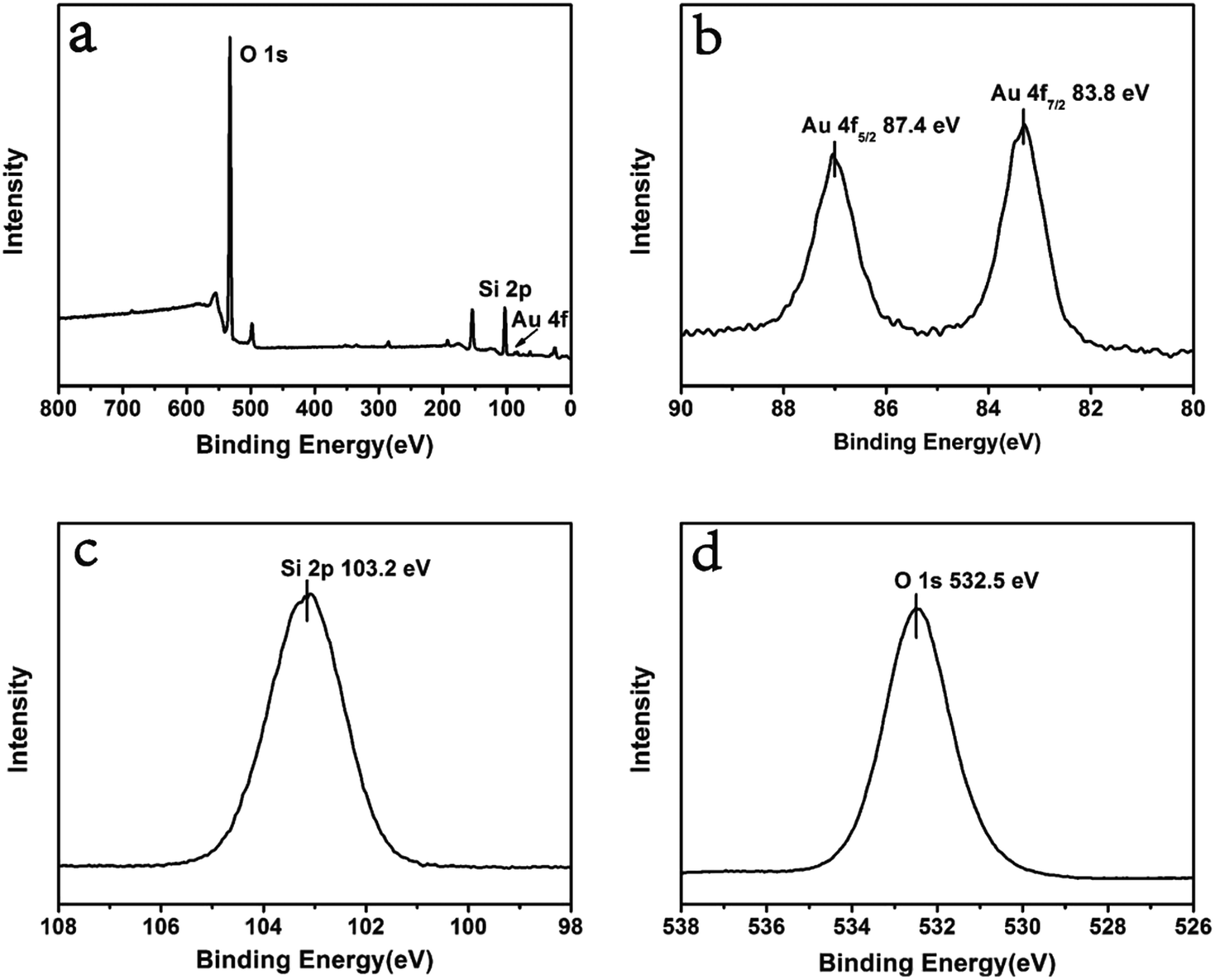 | ||
| Fig. 2 (a) Survey XPS spectrum of ultra small Au–SiO2; (b–d) narrow scan spectra for the elements of Au, Si and O. | ||
XPS analyses were performed to investigate the composition of ultra small Au–SiO2. The results are shown in Fig. 2. O, Si, and Au were observed in the survey spectrum, in agreement with the XRD pattern. There were two peaks in the narrow scan spectrum of Au 4f, with binding energies of 87.4 eV and 83.8 eV, which were attributed to Au 4f5/2 and Au 4f7/2 in Au0, respectively. The single peak of Si 2p with binding energy of 103.2 eV corresponded to the binding state of Si(IV) in SiO2. The O 1s was also a single peak with binding energy of 532.5 eV, corresponding to O(II) in SiO2. These results confirmed that the samples were composed of Au and SiO2.27,28
The porosity of SiO2 was quite indispensable for the planned catalytic application because it made possible the contact of the gold nanoparticles and the reactants. It is worthy of delight that the microemulsion-made SiO2 was reported to be porous.29,30 A nitrogen sorption measurement was carried out to investigate the surface area and pore size of the Au–SiO2. The sorption isotherm (Fig. 1d) showed a microporous feature because there was a sharp capillary condensation process at low pressure. The BET surface area was 99.2 m2 g−1. The size of micropore was calculated by the original Horvath–Kawazoe model in the inset of Fig. 1d, giving the micropore size distribution of 0.55 nm and 0.95 nm. The porosity of the structure makes it possible for Au–SiO2 to be an effective catalyst.31
In the initial stage of the catalytic studies, we selected etherification of dimethylphenylsilane with ethanol as the model reaction. The reaction was carried out using ultra small Au–SiO2 in THF at room temperature under air. As shown in Table 1, dimethylphenylsilane was smoothly converted to ethoxydimethylphenylsilane after 2 hours with over 99% conversion. Furthermore, the only compound observed by GC-MS was the intended product, ethoxydimethylphenylsilane; no disiloxane by-product was detected by GC-MS.
For comparison, gold nanoparticles supported on alternative substrates, Au–Al2O3 and Au–FeOx, were prepared under the same conditions. It was unfortunate that the smallest size of gold nanoparticles obtained on Al2O3 and FeOx were 3.8 nm and 4.0 nm, respectively. To eliminate the impact of the size of the gold nanoparticles, 4.0 nm gold nanoparticles supported on silica were also prepared. The results are shown in Table 1. Certainly, Au (4.0 nm)–SiO2 was also an effective catalyst, although inferior to the ultra small Au–SiO2. This implied that the size of the gold nanoparticles had an impact on the reaction, as had been reported in other reactions.32–34 The gold nanoparticles with the same size but supported on Al2O3 and FeOx afforded lower TOFs. These results suggested that the support also played a key role for enhancing the activity. This is an uncommon outcome because gold nanoparticles supported on silica are generally less active than those supported on metal oxides.35,36
To better understand the differences between these three catalysts, the adsorption of reactants over Au–SiO2, Au–Al2O3 and Au–Fe2O3 was measured (Table S1†). The amount of ethanol adsorbed on Au–SiO2 is 4-fold over that on Au–Al2O3 and Au–FeOx. Even more dramatic, the adsorption amount of PhMe2SiH over Au–SiO2 is 10-fold over the other two supports, reflecting the enriching ability of silica support for both substrates. This substrate enrichment yielded a faster reaction on Au–SiO2 (conversion of PhMe2SiH was 56%) compared to Al2O3 and FeOx (conversion of PhMe2SiH was only 22.2% and 7.5%, respectively). Furthermore, the reaction rate remained unchanged even though ethanol was used as the solvent instead of THF when the reaction was conducted over Au–SiO2, implying that the ethanol was absorbed on the silica to create an ethanol-saturated region around the gold nanoparticles. As a result, the reaction rate had no relation to the concentration of ethanol over Au–SiO2.
To gain more insight into the mechanism, the kinetics were investigated (Fig. 3). The slop of the plot of concentration versus reaction time clearly indicated a faster reaction rate of PhMe2SiH and ethanol on Au–SiO2 versus the same reaction on Au–Al2O3 or Au–FeOx. This was consistent with the favorable adsorption of the substrates on silica. Furthermore, the plot of concentration versus reaction time for Au–SiO2 yielded a straight line for both PhMe2SiH (Fig. 3a, -▲-) and ethanol (Fig. 3b, -▲-). Thus, the reaction rate for Au–SiO2 was independent of the concentration of either PhMe2SiH or ethanol, in contrast to previously reported homogeneous catalysts.37,38 Therefore, the reaction could be considered as zero-order for Au–SiO2 and the surface reaction on gold was could be considered as the rate-determining step.39
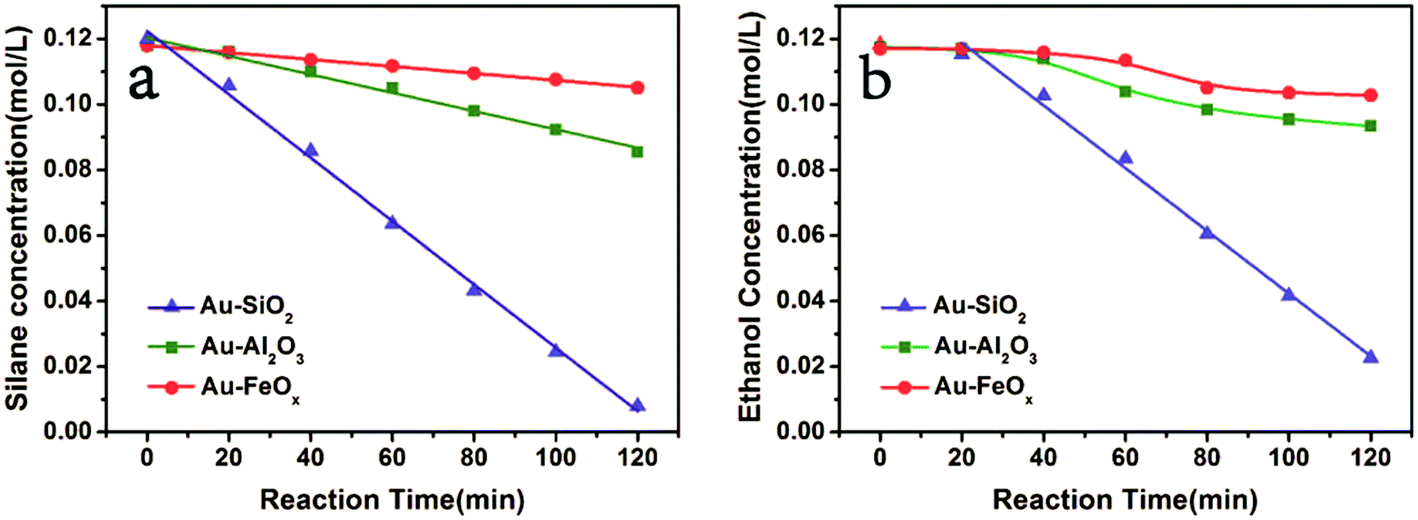 | ||
| Fig. 3 Evolution of the concentration of PhMe2SiH when ethanol is in excess (a) and concentration of ethanol when PhMe2SiH is in excess (b). The reactions were performed at room temperature under air and the conversion was obtained by GC with internal standard. | ||
For Au–Al2O3 and Au–FeOx, the straight lines in Fig. 3a (-■-, -●-) showed that the reaction rate of PhMe2SiH was independent of its concentration (a zero-order reaction). Considering the fact that the amount of adsorption of PhMe2SiH on Au–Al2O3 and Au–FeOx was much smaller than on Au–SiO2, this independence could be attributed to a strong adsorption of Au nanoparticles for PhMe2SiH via interaction with the Si–H bond due to the electron-deficiency of Au nanoparticles.40 Detection of such an interaction in this work was not possible due to the low content of gold in Au–SiO2 (1.37 wt%). Furthermore, the curves in Fig. 3b (-■-, -●-) mean that the reaction rate of ethanol changed with its concentration over Au–Al2O3 or Au–FeOx. Moreover, the slope of the curves decreased gradually as the reaction proceeded, meaning that the reaction rate increased with substrate concentration, which suggested that adsorption of ethanol on Au–Al2O3 or Au–FeOx was the rate-determining step.24 In addition, on account of the strong adsorption of gold nanoparticles for PhMe2SiH and the resulted straight line, a weak adsorption of gold nanoparticles for ethanol could be ascribed. The kinetic analysis of the etherification catalyzed by Au–SiO2 giving a zero order for ethanol was attributed to the enrichment of support-silica.
Both silane and ethanol were collected onto the silica support, affording an enriched substrate layer around the gold nanoparticles and creating the nanoreactors effect. The reactants were adsorbed from the SiO2 onto the surface of the gold nanoparticles, strongly held for PhMe2SiH and weakly held for ethanol. This was followed by initiation of the etherification reaction. The reaction progress could be described by the Langmuir–Hinshelwood model of encounters between two molecular fragments adsorbed on a surface and their consequential reaction.41 A diagrammatic presentation of this process is illustrated in Scheme 1. As the reaction followed a zero order mechanism, it is worth mentioning that the surface etherification process was the rate-determining step for the whole reaction progress.39
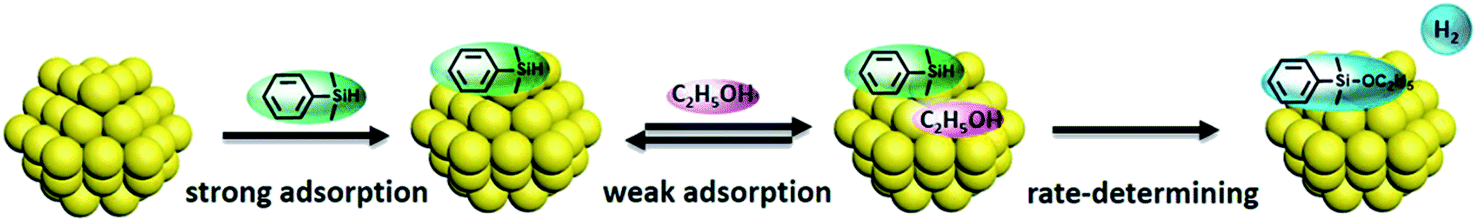 | ||
| Scheme 1 Reaction progress for the etherification on gold nanoparticles. | ||
Reusability is an important feature of heterogeneous catalysts that is superior to homogenous ones. To assess the reusability of the ultra small Au–SiO2, a series of ten consecutive runs of the etherification were carried out. Recycling experiments proceeded without loss of activity or selectivity, and all the runs could be finished in 2 hours with almost 99% conversion (Table S2†). After ten runs, the TEM images (Fig. S8†) showed no alteration of the catalyst morphology and no aggregation of gold nanoparticles during the reuse. Moreover, the content of gold ions in the solution was only 0.0613 mg L−1 as detected by ICP, which illustrated that there was no outflow of gold (45.7 mg L−1 addition). Certainly, the ultra small Au–SiO2 catalyst has excellent reusability.
The scope of the Au–SiO2-catalyzed etherification was then investigated on a panel of silanes and alcohols (Table 2). The different substrates could react not only with ethanol but also with other alcohols, such as methanol, n-butanol and benzyl alcohol, under mild conditions, producing the corresponding alkoxysilanes in excellent yields (all the conversions were higher than 90% and most of them were 99%). A comparison between our study and the recently reported literature is given in Table S3;† the results indicated that the ultra small Au–SiO2 was an active and effective catalyst. More importantly, all the reaction proceeded cleanly with no disiloxane formation. It was noted that shorter chain alcohols such as methanol showed higher reactivity in the synthesis. Higher temperatures were required for large alcohols such as n-butanol (50 °C) and benzyl alcohol (80 °C). Reactions of large silanes encountered the same situation; for example, etherification of Ph2MeSiH with benzyl alcohol yielded only 5%. This may be ascribed to the limitations due to steric hindrance.40,42
Conclusion
In summary, we successfully developed ultra small gold nanoparticles supported on porous silica (Au–SiO2), and applied the samples as catalysts for the etherification of silanes with efficient activity, high selectivity, and reusability. Favorable enrichment of both PhMe2SiH and ethanol on silica was observed, which allowed the Au–SiO2 to function as nanoreactors with saturated reactant layers around the gold nanoparticles. The reaction progress was initiated following the Langmuir–Hinshelwood model, featuring strong adsorption of PhMe2SiH and weak adsorption of ethanol on gold nanoparticles. The rate-determining step was initiation of the etherification process on the gold surface. The reaction kinetics were studied and the results indicated that the reaction catalyzed by Au–SiO2 followed a zero order, which was different than previously reported homogeneous catalysts and heterogenous Au–Al2O3 and Au–FeOx catalysts prepared by the same method.Acknowledgements
This work thanks the financial support from the National Natural Science Foundation of China (Grant No. 21421005, 21576044 and 21536002), the Fundamental Research Foundation of Liaoning Ministry of Education (Grant No. LZ2015020), the Fundamental Research Funds for the Central Universities (No. DUT15ZD224) and Dalian University of Technology Innovation Team (DUT2016TB12).References
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469–4506 CrossRef CAS PubMed.
- V. Dhanala, S. K. Maity and D. Shee, RSC Adv., 2015, 5, 52522–52532 RSC.
- M. A. Newton, Chem. Soc. Rev., 2008, 37, 2644–2657 RSC.
- P. Munusamy, S. Sanghavi, T. Varga and T. Suntharampillai, RSC Adv., 2014, 4, 8421 RSC.
- M. Opanasenko, P. Štěpnička and J. Čejka, RSC Adv., 2014, 4, 65137–65162 RSC.
- U. Jeong, J. B. Joo and Y. Kim, RSC Adv., 2015, 5, 55608–55618 RSC.
- X. Liu, L. He, Y. M. Liu and Y. Cao, Acc. Chem. Res., 2014, 47, 793–804 CrossRef CAS PubMed.
- N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC.
- K. Karki, A. Materny and D. Roccatano, Phys. Chem. Chem. Phys., 2011, 13, 11864–11871 RSC.
- T. W. G. P. G. M. Wuts, Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Hoboken NJ, 4th edn, 2007, p. 167 Search PubMed.
- Z. Li, S. Lin, L. Ji, Z. Zhang, X. Zhang and Y. Ding, Catal. Sci. Technol., 2014, 4, 1734 CAS.
- J. F. Blandez, A. Primo, M. Alvaro and H. Garcia, Angew. Chem., Int. Ed. Engl., 2014, 53, 12581–12586 CAS.
- Z. Li, C. Zhang, J. Tian, Z. Zhang, X. Zhang and Y. Ding, Catal. Commun., 2014, 53, 53–56 CrossRef CAS.
- P. Raffa, C. Evangelisti, G. Vitulli and P. Salvadori, Tetrahedron Lett., 2008, 49, 3221–3224 CrossRef CAS.
- T. Taguchi, K. Isozaki and K. Miki, Adv. Mater., 2012, 24, 6462–6467 CrossRef CAS PubMed.
- S. H. Petrosko, R. Johnson, H. White and C. A. Mirkin, J. Am. Chem. Soc., 2016, 138, 7443–7445 CrossRef CAS PubMed.
- R. Ciriminna, A. Fidalgo, V. Pandarus, F. Beland, L. M. Ilharco and M. Pagliaro, Chem. Rev., 2013, 113, 6592–6620 CrossRef CAS PubMed.
- A. Rimola, D. Costa, M. Sodupe, J. F. Lambert and P. Ugliengo, Chem. Rev., 2013, 113, 4216–4313 CrossRef CAS PubMed.
- B. Coasne, A. Galarneau, R. J. Pellenq and F. Di Renzo, Chem. Soc. Rev., 2013, 42, 4141–4171 RSC.
- K. A. Dahlberg and J. W. Schwank, Chem. Mater., 2012, 24, 2635–2644 CrossRef CAS.
- H. Sun, Q. Tang, Y. Du, X. Liu, Y. Chen and Y. Yang, J. Colloid Interface Sci., 2009, 333, 317–323 CrossRef CAS PubMed.
- E. L. Margelefsky, R. K. Zeidan and M. E. Davis, Chem. Soc. Rev., 2008, 37, 1118–1126 RSC.
- W. Li, A. Wang, X. Yang, Y. Huang and T. Zhang, Chem. Commun., 2012, 48, 9183–9185 RSC.
- G. S. B. Corain and N. Toshima, Metal nanoclusters in catalysis and materials science : the issue of size control, Linacre House, Jordan Hill, Oxford OX2 8DP, UK, 2008, pp. 6–8 Search PubMed.
- Z. Wu, J. Chen and R. Jin, Adv. Funct. Mater., 2011, 21, 177–183 CrossRef CAS.
- T. Wang, X. Yuan, S. Li, L. Zeng and J. Gong, Nanoscale, 2015, 7, 7593–7602 RSC.
- A. S. Sharma, H. Kaur and D. Shah, RSC Adv., 2016, 6, 28688–28727 RSC.
- H. Yamauchi, T. Ishikawa and S. Kondo, Colloids Surf., 1989, 37, 71–80 CrossRef.
- J. Wang, X. Li, S. Zhang and R. Lu, Nanoscale, 2013, 5, 4823–4828 RSC.
- D. Weber, A. J. Sederman, M. D. Mantle, J. Mitchell and L. F. Gladden, Phys. Chem. Chem. Phys., 2010, 12, 2619–2624 RSC.
- T. Zhang, H. Zhao, S. He, K. Liu, H. Liu, Y. Yin and C. Gao, ACS Nano, 2014, 8, 7297–7304 CrossRef CAS PubMed.
- L. Francois, M. Mostafavi, J. Belloni and J. A. Delaire, Phys. Chem. Chem. Phys., 2001, 3, 4965–4971 RSC.
- A. Ahmad, Y. Wei, F. Syed, M. Imran, Z. U. H. Khan, K. Tahir, A. U. Khan, M. Raza, Q. Khan and Q. Yuan, RSC Adv., 2015, 5, 99364–99377 RSC.
- M. S. Chen and D. W. Goodman, Science, 2004, 306, 252–255 CrossRef CAS PubMed.
- A. Taketoshi and M. Haruta, Chem. Lett., 2014, 43, 380–387 CrossRef CAS.
- M. Yu, H. Jing and X. Fu, Inorg. Chem., 2013, 52, 10741–10743 CrossRef CAS PubMed.
- D. Gao and C. Cui, Chem.–Eur. J., 2013, 19, 11143–11147 CrossRef CAS PubMed.
- J. D. P. Peter Atkins, Atkins' Physical Chemistry, Oxford University Press, 2006, p. 796 Search PubMed.
- T. Mitsudome, Y. Yamamoto, A. Noujima and K. Kaneda, Chem.–Eur. J., 2013, 19, 14398–14402 CrossRef CAS PubMed.
- J. D. P. Peter Atkins, Atkins' Physical Chemistry, Oxford University Press, 2006, p. 927 Search PubMed.
- A. Dhakshinamoorthy, P. Concepcion and H. Garcia, Chem. Commun., 2016, 52, 2725–2728 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra22359a |
| This journal is © The Royal Society of Chemistry 2016 |