
Promising ESIPT-based fluorescence sensor for Cu2+ and CN− ions: investigation towards logic gate behaviour, anticancer activities and bioimaging application†
Abstract
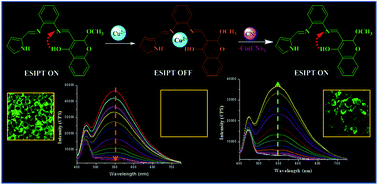
An excited state intramolecular proton transfer (ESIPT) process-based novel chromogenic and fluorogenic probe (2) was synthesized with the aim of sequential in situ detection of Cu2+ and CN− ions under aqueous and biological conditions. The probe revealed chelating affinity for transition metal cations, amidst which only Cu2+ efficiently quenches the emission intensity. Further in situ addition of CN− results in a metal displacement reaction and a turn on fluorescence response. This quencher displacement sensing strategy results in “ON–OFF–ON” type of fluorescence changes with high selectivity and great affinity in nanomolar detection of [CN−] and outlines the working principle of the IMPLICATION logic gate. The in vitro cytotoxic activity studied via the MTT assay revealed that in situ-formed Cu complexes worked as potential anticancer chemotherapeutic agents towards MCF-7 (human breast adenocarcinoma) cell lines (2 + Cu2+ IC50 = 5.69 ± 0.26 μg mL−1, 3 + Cu2+ IC50 = 7.36 ± 0.29 μg mL−1). The selective detection of Cu2+ and CN− ions in biological systems was also explained by intracellular bioimaging studies in MCF-7 cell lines.
 Please wait while we load your content...
Please wait while we load your content...

