
Effect of Ni substitution on structural stability, micromorphology, and electrochemical performance of Li2MnSiO4/C cathode materials
Hui Dengab,
Shi-Xi Zhao*a,
Xia Wuab,
Lei Weiab,
Yu-Feng Denga and
Ce-Wen Nanb
aGraduate School at Shenzhen, Tsinghua University, Shenzhen, 518055, China. E-mail: zhaosx@sz.tsinghua.edu.cn; Fax: +86 0755 26036372; Tel: +86 0755 26036372
bSchool of Materials Science and Engineering, Tsinghua University, Beijing, 100084, China
First published on 14th November 2016
Abstract
The chief drawback of Li2MnSiO4 cathode materials is structural instability deriving from the transformation of [MnO4] tetrahedra to [MnO6] octahedra during oxidation from Mn(II) to Mn(III) and the Jahn–Teller effect especially from Mn(III) to Mn(IV). Based on the theory that Ni2+ could keep the [NiO4] ligand unchanged and stable during oxidation/reduction, [NiO4] tetrahedra may behave as a regional structural framework to support the total crystal lattice and alleviate structural distortion partially. In this paper we synthesize nano-Li2Mn1−xNixSiO4/C samples with a small amount of Ni as the dopant via a hydrothermal route. Firstly, we discuss the existential state of Ni through XPS spectra, and demonstrate that both Ni2+ and metallic Ni coexist, especially Ni2+ could improve structural stability. The influence of Ni doping on morphology, structural stability, and electrochemical performance is discussed through Raman spectroscopy and electrochemical tests below. It is confirmed that there exists an optimal Ni content under which the optimal performance is displayed. In our work, the optimum Ni content is 5.0%, and the maximal discharge capacity achieved is 235 mA h g−1 for the 5.0% Ni doped sample in the first cycle.
1. Introduction
The urgency of the energy crisis and environmental pollution makes lithium-ion batteries increasingly important, especially in providing power for energy storage systems such as hybrid electric vehicles and portable electronic devices, so developing superior cathode materials is crucial in future research.1 Compared with traditional lithium-ion intercalated transition metal oxides (e.g. LiCoO2, LiMn2O4), polyanionic compounds have remarkable advantages such as high capacity, safety and thermal stability because of strong covalent bonds between nonmetallic atoms.2–4 Among the series of polyanionic compounds (LixMy(XOn)z, M = Fe, Mn, Co, X = P, Si), Li2MnSiO4 has been proposed as the most potential material because it theoretically extracts two lithium ions totally and has a reversible capacity above 330 mA h g−1.5–7 It is hard for Li2FeSiO4 and Li2CoSiO4 to extract two lithium ions,8,9 because the oxidation voltage is predicted to be outside the voltage stability window of common electrolytes.10 Furthermore, abundant resource, low cost and non-toxic precursors are also essential factors for the possible application of Li2MnSiO4. Besides, Li2MnSiO4 exhibits four types of structural forms, including two orthorhombic phases (Pmnb, Pmn21) and two monoclinic phases (P21/n, Pn), separately related to either β or γ Li3PO4 polymorphs.11–14 The current research focuses more on orthorhombic Li2MnSiO4 polymorphs because of their higher potential and commercial value, and the object of our study is the Pmn21 polymorph.Despite the promising prospect in theory, a lot of theoretical and technical problems have not been resolved still such as, poor cycling stability, reversibility and actual low discharge capacity etc. The most crucial drawback of this material is its structural instability during charge/discharge process.11–16 During the oxidation of Mn2+ to Mn3+, the structural collapse is mainly derived from the transformation of [MnO4] tetrahedron into [MnO6] octahedron. Furthermore the dominant variation during the next oxidation of Mn3+ to Mn4+ is the Jahn–Teller effect, under which the [MnO6] octahedron would be lengthened along the center axis, causing more obvious volume distortion and worse symmetry, even though the coordination number remains unchanged. That's why the material probably turns into amorphous state because of collapsing structure, the lithium-ion can't be extracted totally and the actual capacity descends promptly in initial cycles. Another dominating drawback of this material is the extremely low electronic conductivity (only 10−16 S cm−1 at room temperature) since [MnO4] tetrahedrons are surrounded by insulating [SiO4] tetrahedrons. So carbon coating is a usual method to improve electronic conductivity.17–20 In addition, the polarization in charge/discharge process, Li–Mn anti-site defect and dissolution in electrolyte are also negative factors to influence the performance of Li2MnSiO4.
In most cases, the modification has been completed through cationic doping based on the theory of inhibiting the Jahn–Teller effect via adjusting electronic distribution of transition metal ionic d-orbitals and optimizing the geometry configuration, or increasing the lithium-ion content via defect reactions so as to raise initial discharge capacity.21–27 However the intrinsic problem of structural collapse and drastic capacity fading still exists, which is closely related with the reversibility and capacity retention, so it's key to raise the capacity retention rather than increase the discharge capacity of initial cycles, because the actual discharge capacity of modified Li2MnSiO4 in first few cycles has already been higher than some commercial cathodes. Therefore searching an approach to stabilize the preliminary high capacity is important in prospective research.
Based on crystal field theory, some researchers try to introduce some kind of cation that could keep ligand unchanged and stable during oxidation, these stable ligands as structural framework to support the crystal lattice and prevent or ameliorate the regional structural degradation. Based on this, some research tried to use ferric salt or nickel salt as precursors. Ruiyong Chen et al. synthesized Li2Fe1−yMnySiO4 nanocrystalline and the results showed better retention than pure Li2MnSiO4 but still poor reversibility to date.28 However, the Fe2+ dopant also leads to theoretical capacity reduction because the redox reaction (Fe3+/Fe4+) is impractical.29–31 While nickel salt is relatively better choice because the redox reaction (Ni2+/Ni4+) can take place theoretically, and substituting Ni2+ having close radius with Mn2+ would not cause the crystalline distorted excessively. The DFT calculations in A. Saracibar's research also suggest that Mn substituted by Ni might help to prevent the structural collapse because when Li2MnSiO4 is doped with transition metal ion (Mg2+, Fe2+, Co2+, Ni2+), Ni2+ is investigated as the only one which does not produce octahedral Mn4+ in any delithiated Li1−yMn1−xNixSiO4 configuration.32 But the current study about Ni2+ doping is rare and the previous results show it's not easy to prepare Ni2+ doping sample because the reducing conditions of the carbon coating process (high temperature heating in Ar) easily form Ni nanoparticles.32,33 In this work, we choose Ni2+ as dopant and synthesize the samples with different content of Ni element. Not only the existential valence state of Ni element has been firstly discussed, the influence of Ni element on the crystalline, and whether the expected stability effect of Ni2+ really works in structural transformation and capacity degradation has been also discussed.
2. Experimental
2.1 Synthesis of Li2MnSiO4/C nanoparticles
Samples of Li2MnSiO4/C nanoparticles were synthesized through a hydrothermal route. Tetraethyl orthosilicate (TEOS) (Aladdin, 99%) and Mn(Ac)2·H2O (Aladdin, 99%) were mixed in solvents of ethanol, distilled water and NH3·H2O (volume ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1) for several hours in order to be fully hydrolyzed. Then the admixture was kept at 150 °C for 24 h in Teflon-lined stainless steel autoclave and the intermediate compounds were mixed homogeneously with Li2CO3 (Aladdin, AR) through the ball milling method. After the mixture was calcined at 700 °C for 10 h under Ar/H2 (95/5 vol%) atmosphere in tube furnace, the product obtained as pristine Li2MnSiO4.
:2:1) for several hours in order to be fully hydrolyzed. Then the admixture was kept at 150 °C for 24 h in Teflon-lined stainless steel autoclave and the intermediate compounds were mixed homogeneously with Li2CO3 (Aladdin, AR) through the ball milling method. After the mixture was calcined at 700 °C for 10 h under Ar/H2 (95/5 vol%) atmosphere in tube furnace, the product obtained as pristine Li2MnSiO4.
Starch ((C6H10O5)n, Aladdin, AR) as carbon source was divided into two parts to add. The first one was homogeneously mixed with admixture and stirred for several hours before hydrothermal reaction. Because a small quantity of carbon added during hydrothermal process can inhibit grain growth, particle agglomeration and accelerating reaction uniformity. Then the second part of starch was heated in about 70 °C water bath for several hours to be fully gelatinized, through which the starch would swell to many times of common volume, so that the gelatinized starch could coat on the surface of particles more homogeneously.
For discussing the influence of different Ni content, Ni(Ac)2·H2O (Aladdin, 99%) was added in calculated quantity to offer corresponding partial Ni2+. As a comparison, Li2Mn1−xNixSiO4/C (x = 0, 2.5%, 5.0%, 7.5%) composites were synthesized in our experiment. They are named respectively as pristine Li2MnSiO4/C, 2.5% Ni doped sample, 5.0% Ni doped sample and 7.5% Ni doped sample.
2.2 Structural characterizations
X-ray diffraction patterns of the synthesized powders were collected by X-ray diffraction (XRD, Rint-2000V/PC, Rigaku, Japan) using Cu-Kα radiation (l = 0.154 nm) and θ/2θ Bragg Brentano geometry. The data were recorded in the range of 10–60° at a scanning rate of 5° per min. The particle morphology was observed with scanning electron microscope (SEM, Carl Zeiss, ZEISS SUPRA®55) and high resolution transmission electron microscope (HRTEM, JEOL-2011). Laser Raman Tester (HORIBA, LabRam HR800) was used to reflect the structural changes of active substance during charge/discharge process, and X-ray photoelectron spectra (Thermo Fisher, ESCALAB 250X) was applied to measure existential state of elements. The carbon content of the products was tested by thermal gravimetric-differential scanning calorimetry analysis (NETZSCH STA 449F3 thermal analyzer) in air from room temperature to 900 °C at a heating rate of 10 °C min−1.2.3 Cells assembly and electrochemical tests
To fabricate the cathode of a cell, the 80 wt% active material need to be mixed with 10 wt% carbon black and 10 wt% polyvinylidene fluoride (PVDF) dispersed in N-methylpyrrolidone (NMP) and stirred for several hours to obtain the slurry. The slurry was coated onto aluminum foil and then dried at 120 °C for 10 h in a vacuum oven. The coated foil was about 120 μm thick and cut into circular discs, 15 mm in diameter. The lithium foil was used as anode, and 1 M LiPF6 in ethylene carbonate (EC)–dimethyl carbonate (DMC) mixture (volume ratio of 1:1) was used as the electrolyte solution, and Celgard 2300 polypropylene film as the separator. All of them were assembled as coin cells in an argon-filled glove box (H2O, O2 ≤ 0.5 ppm).
The charge/discharge cycles were carried out using a LAND CT2001A tester at a rate of 0.05C in the range of 1.5–4.8 V at 25 °C. And the electrochemical impedance spectroscopy was examined by applying an AC voltage of 5 mV over the frequency range of 0.01 Hz to 100 kHz in the same workstation.
3. Results and discussion
3.1 Structural analysis
In this work, Pmn21 polymorph is prepared, and Ni2+ substitute for partial Mn2+ in modified materials. The possible crystal structures of the prepared Li2MnSiO4 and Li2Mn1−xNixSiO4 are as shown in Fig. 1, wherein Ni2+ occupies and replaces Mn2+ partially, forming [NiO4] tetrahedrons, which substitute for a few of [MnO4] tetrahedrons.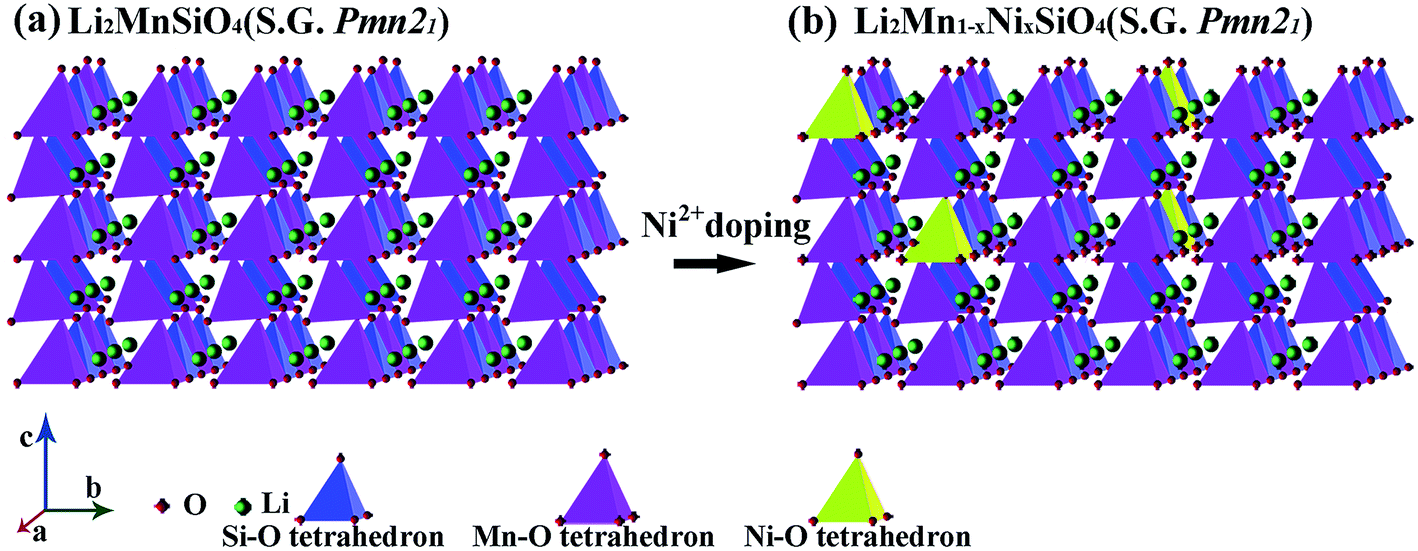 | ||
| Fig. 1 The crystal structures (a) Li2MnSiO4 (b) Li2Mn1−xNixSiO4. | ||
The fact that phase structure of Li2Mn1−xNixSiO4 keeps unchanged is proved by XRD patterns, which of the pristine and modified Li2MnSiO4/C samples are as shown in Fig. 2. The peaks are all in good agreement with orthorhombic phase of Pmn21 space group, and the indices of typical crystal plane have been marked.34,35 It means Ni element doping has no influence on main crystalline structure, but the diffraction peaks of modified samples become broader, the possible explanation is lower crystallinity of the sample, or smaller crystallite size, and the latter has been proved by the corresponding SEM and TEM figures. It's inevitable to form some impurities such as MnO and Mn2SiO4 in as-prepared samples via hydrothermal route.11 As the increase of Ni element content, the relative impurities decrease, so we assume Ni2+ doping is favorable in inhibiting the formation of impurities, but the new MnO peak emerges in 7.5% Ni doped sample. Referring to the standard Pmn21-Li2MnSiO4 XRD pattern, the lattice parameters are determined by GASA refinement, as listed in Table 1. The substitution of Ni element leads to the continuous increasing of the lattice constant b and c, which kind of variation probably derives from larger radius of doped Ni2+(0.69 a) compared to Mn2+(0.67 Å).
 | ||
| Fig. 2 The XRD patterns of the pristine Li2MnSiO4/C and modified samples. | ||
| x | a (Å) | b (Å) | c (Å) | Rwp (%) | Rp (%) |
|---|---|---|---|---|---|
| 0 | 6.3000(1) | 5.3737(5) | 4.9697(5) | 12.8 | 10.2 |
| 2.5% | 6.2994(1) | 5.3798(9) | 4.9721(5) | 11.0 | 8.6 |
| 5.0% | 6.2980(2) | 5.3798(7) | 4.9734(3) | 12.6 | 9.3 |
| 7.5% | 6.2965(7) | 5.3864(4) | 4.9751(6) | 14.3 | 10.1 |
Besides, the right part of Fig. 2 is enlarged part of XRD pattern in the range of 43° to 53°, it proves the existence of metallic Ni unavoidably, the peaks of which are easily covered up by the peaks of (220) and (122), and the precipitation as Ni nanoparticles could explain the increase of MnO in high Ni element content doped sample.32,33 To exclude the potential reducing effect of trace H2 in sintering atmosphere, the high pure N2 is applied, which still causes the production of metallic Ni. The reduction conditions are most likely to be derived from the carbonization of the cladding layer. We preliminarily conclude the coexistence of Ni2+ and metallic Ni, which is also verified by the following tests. Finally no peaks corresponding to carbon have been observed here, which indicates the embedding carbon is amorphous.
3.2 Existential state of Ni atom
To fully certify the existence of bivalent nickel ion, the X-ray photoelectron spectroscopy is applied to analyze the valence state of nickel element. Fig. 3(a) and (b) display the atom hybridization state of Mn and Si in pure sample and modified samples separately, where the binding energy of Mn 2p3/2 (641.7 eV) is corresponding to Mn2+, showing that the divalent state of manganese in our samples.17,36 And it is almost unchanged with Ni element substitution content, and accords well with past results.32 According to the basically conformity of fitting consequences, we can conclude that the doping of Ni element has no much effect on hybridization state of manganese element and silicon element. Fig. 3(c) illustrates that the atom hybridization state of nickel ion is 2p orbital hybridization, and it has three peaks, with the binding energy in 854.5 eV, 860.4 eV and 879.1 eV respectively through accurate fitting. A. Saracibar's work shows Ni 2p spectra do not change at all with charge voltage most likely due to its precipitation as metallic Ni nanoparticles, which only exhibits peak in 854.5 eV.32 This peak is in good agreement with our results, and the extra 879.1 eV and 860.4 eV peaks correspond to different hybridization state of divalent nickel ion, which verify the coexistence of Ni2+ and metallic Ni. And along with the increase of Ni element content, the ratio of two forms is almost same. Above all, the existence of Ni2+ and the redox of Ni2+ have been proved by XPS.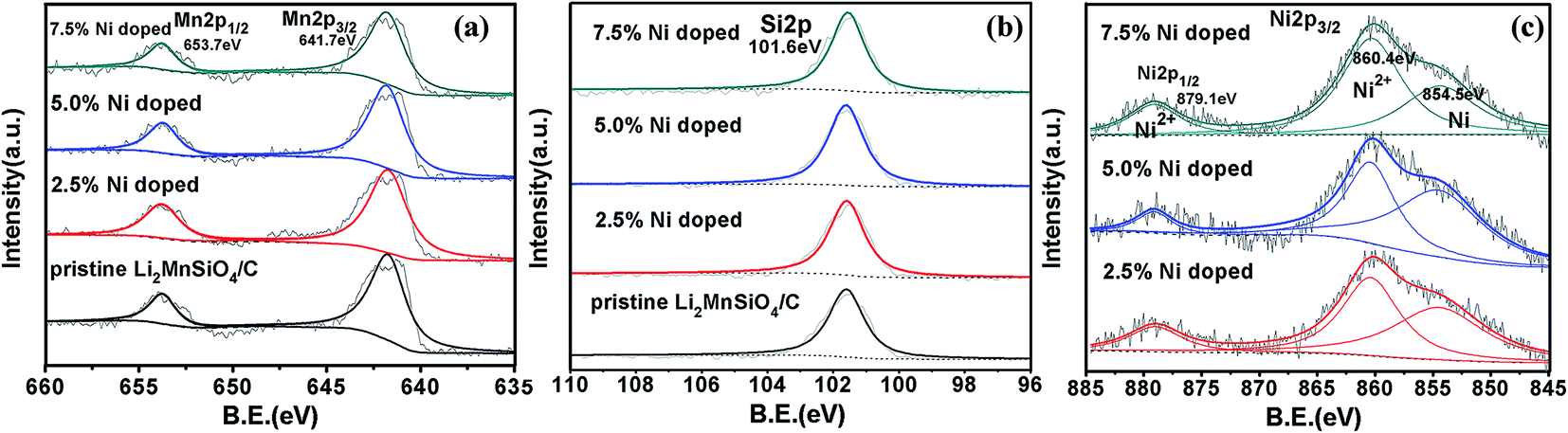 | ||
| Fig. 3 Ex situ XPS patterns of the samples in pristine state (a) The Mn 2p spectra (b) The Si 2p spectra (c) The Ni 2p spectra in modified materials. | ||
3.3 Effect of grain refinement
Except for doping and carbon coating, nanocrystallization of grains is also a main task of material modification, the most remarkable advantage of which is increasing contact area between active materials and conductive material, in order to shorten the diffusion distance of lithium ion and further facilitate transport of lithium-ion via those densely packed nano-sized grains. Besides the conductive carbon coating connects the nanoparticles in close crystal domains, providing highly conductive channels for electron mobility between neighboring Li2MnSiO4 nanoparticles.23 So the electrochemical performance of samples with nanoparticles micromorphology would be improved. In this paper, we focus on the grain refinement effect, the possible explanation of which is the Ni element doping.Fig. 4(a)–(d) are the SEM images of the pristine Li2MnSiO4/C, Li2Mn97.5%Ni2.5%SiO4/C, Li2Mn95.0%Ni5.0%SiO4/C and Li2Mn92.5%Ni7.5%SiO4/C respectively, where the grain refinement phenomenon emerges apparently. As shown in Fig. 4, Ni element substitution is effective in reducing the grain size, and grain size is homogeneous, no other abundant impurities are observed. The fact is much more obvious in TEM images. Fig. 4(e) is the TEM image of pristine Li2MnSiO4/C, Fig. 4(f)–(h) correspond to the TEM images of the modified sample with 2.5%, 5.0% and 7.5% Ni element substituted. The particles in Fig. 4(e) are inhomogeneous, the size range covers from about 30 nm to over 100 nm and the carbon layer is thick. However Fig. 4(f) distinctly exhibits the homogeneity of arrayed particles in visual field, the average size of which is estimated about 20 nm, and the carbon layer is thinner and more uniform. The possible explanation of grain refinement effect is the decrease of surface energy, so as to prevent grain growth because of doping.37 The similar morphology appears in Fig. 4(g) and (h), but particles in Fig. 4(h) occur agglomeration.
 | ||
| Fig. 4 The SEM images of the nanocomposites (a) Li2MnSiO4/C (b) Li2Mn97.5%Ni2.5%SiO4/C (c) Li2Mn95.0%Ni5.0%SiO4/C (d) Li2Mn92.5%Ni7.5%SiO4/C and the TEM images of the materials (e) Li2MnSiO4/C and (f) Li2Mn97.5%Ni2.5%SiO4 (the insert is enlarged figure of one particle), (g) Li2Mn95.0%Ni5.0%SiO4 and (h) Li2Mn92.5%Ni7.5%SiO4. | ||
Besides, nanoparticles with less than 20 nm in diameter would provide shorter pathways for quick lithium-ion and electronic conduction within the nanoparticles.38 The inserted picture in Fig. 4(f) also suggests the crystal fringes and coating carbon layer of an intact grain, the fringe spacing corresponding to (210) plane. The improvement in transport of lithium-ion and electric conductivity originated from the grain refinement may be one of the most important factors to influence the electrochemical performance.
3.4 Improvement in structural stabilization
Raman tester is used to characterize structural stability of active substance to a certain degree, which reflects the capacity retention during charge/discharge process. According to the previous reports, the peak around 615 cm−1 is assigned to the symmetric Mn–O stretching vibration of [MnO4] group and the strong band around 635 cm−1 is assigned to the symmetric Mn–O stretching mode of [MnO6] octahedron (A1g).39–42 When the oxidation reaction occurs, the characteristic peak of Mn–O bonds in laser Raman spectrograms would shift right. The previous results show even only after first cycle, the Raman shift has shifted right obviously from 619 cm−1 to 650 cm−1, this transformation can reflect the rapid variation of [MnOx] ligand in Li2MnSiO4.40 On the other hand, the peak around 490 cm−1 is associated with the Ni2+–O stretching mode in the structure and the peak located in 370 cm−1 could be assigned to the first-order transverse optical mode of Ni2+–O.43,44 And no Raman peak corresponding to pure metallic Ni would appear in figures.In this work, we test the Raman spectrograms of a series of samples doped with different Ni element content in the range of 100–2000 cm−1, as shown in Fig. 5(a). Along with the increase of Ni element content, the characteristic peak of [MnO4] ligand keeps unchanged basically, but the intensity of Ni2+–O peaks which are detected in 370 cm−1 and 490 cm−1 gradually increases, representing that the quantity of [NiO4] tetrahedrons and Ni2+–O bonds has kept rising. Complementally, the peaks of D band and G band in diagrams reveal the existing forms of C–C bonds, which composed of both disordered carbon and ordered carbon.29,38
 | ||
| Fig. 5 The Raman spectra of (a) as-prepared Li2Mn1−xNixSiO4/C samples with different Ni content and samples before 1st cycle and after 1st cycle (b) pristine Li2MnSiO4/C (c) Li2Mn97.5%Ni2.5%SiO4/C (d) Li2Mn95.0%Ni5.0%SiO4/C (e) Li2Mn97.5%Ni2.5%SiO4/C (the inserted figures correspond to enlarged parts of [MnOx] transformation). | ||
To analyze the degree of structural distortion according to the Raman results. The variation of Mn–O bonds of the pristine sample and the Ni-doped samples were tested before cycling and after first cycle. Fig. 5(b) is the spectra of the pristine Li2MnSiO4/C, and Fig. 5(c)–(e) are the spectra of the Ni-doped samples. Refer to the immobile peaks of C–C bonds, the right shift of Mn–O bonds from 615 cm−1 to 642 cm−1 is obvious. In comparison, the Raman shift of Mn–O bonds in the 2.5% Ni doped sample only occurs in the range of 615 cm−1 to 625 cm−1 after first cycle, besides 620 cm−1 for 5.0% Ni doped sample and 626 cm−1 for 7.5% Ni doped sample respectively, which reflects smaller part of [MnO4] tetrahedrons in modified samples falling into structural degradation, and the distortion extent of them is much lower than the structure of pristine sample. Especially the 5.0% Ni doped sample also displays lowest distortion extent. As presumed, Ni substitution does effectively lower the structural distortion extent, most likely because of Ni2+ acting as a pillar to prevent lattice collapsing. And for Ni–O bond, the peak position always keeps unchanged in 490 cm−1, which means the coordination number of Ni2+ is unchanged.
3.5 Analysis of electrochemical performance
Previous DFT results predict that Li2NiSiO4 has high voltages platform, 4.5 V for the Ni2+/Ni3+ couple and 5.2 V for the Ni3+/Ni4+ couple.32 We can thus assume that Ni element substitution would increase the average voltage platform of Li2MnSiO4. Fig. 6(a) displays the galvanostatic charge/discharge curves of first cycle of pristine Li2MnSiO4/C and Li2Mn1−xNixSiO4/C nanocomposites, and discharge platforms increase for the modified samples as predicted. Thus the modified materials reveal improved capacity and higher discharge voltage, especially 5.0% Ni doped sample owns highest capacity and voltage platform. On the other hand the reduced capacity in 7.5% Ni doped sample probably results from the influence of metallic Ni. | ||
| Fig. 6 (a) The charge and discharge curves of the as-prepared Li2Mn1−xNixSiO4/C nanocomposites (b) the midpoint potential curves of the as-prepared Li2Mn1−xNixSiO4/C nanocomposites (cycled at room temperature, the rate is 0.05C, the voltage range is 1.5–4.8 V). | ||
The discharge midpoint potential curves of samples are obtained in Fig. 6(b), the same tendency of midpoint voltage accords with Fig. 6(a) to some extent. Moreover, it also conforms to the theoretical trend of increasing average voltage, suggesting a new redox reaction with higher oxidation voltage exists. We infer that partial Ni2+ enters into crystal lattice and is oxidized indeed, as mentioned above. However, along with the much higher Ni element content, a peak value of potential appears in 5.0% Ni doped sample. The possible reason is the limitation of Ni2+ substitution and oxidation in lattice, that's why 5.0% Ni doped sample displays best performance and there is no obvious midpoint potential difference between 5.0% doped sample and 7.5% doped sample as shown in Fig. 6(b). We think there is an optimal balance quantity between metallic Ni/Ni2+ that would has a best improvement effect on electrochemical performance, but once the metallic Ni content is over some limit, the performance may deteriorate instead.
Fig. 7(a) exhibits the cycle curves of Li2MnSiO4/C and Li2Mn1−xNixSiO4/C nanocomposites at room temperature, which reflects the discharge capacity results and the electrochemical retention performance. The raised capacity of modified samples is consistent with Fig. 6(a) especially in preliminary cycles, and the maximal discharge capacity in first cycle could achieve above 235 mA h g−1 within 5.0% Ni doped sample. Although the capacity degradation phenomenon still exists, the descending trend has been weakened. The raised capacity mainly results from the stabilization of Ni2+ and the reduced resistivity caused by metallic Ni.
 | ||
| Fig. 7 (a) The discharge capacity curves of the as-prepared Li2Mn1−xNixSiO4/C nanocomposites (cycled at room temperature, the rate is 0.05C, the voltage range is 1.5–4.8 V). And the calculated capacity retention is as shown in (b). | ||
Fig. 7(b) displays the specific calculated capacity retention results based on the second discharge capacity as the initial capacity because of the possible inaccuracy of first cycle. It's noticeable the Ni element doped materials also show improved retention in initial several cycles. The curves show similar superiority among modified samples more visually, but the superiority disappears after 30 cycles. Associated with the Raman spectroscopy, the alleviated fading capacity exactly accords with the lower distortion extent of structure. Therefore substituted Ni2+ not only promotes the initial discharge capacity but also alleviates the capacity decay in preliminary stage of cycles via acting as a pillar to prevent regional lattice collapsing. Besides, among the samples with different Ni element content, the similar improvement effect in retention may imply that no matter the Ni element dopant amount, the effective Ni2+ as [NiO4] is finite, as suggested from Fig. 6(b). But the promotion is still weak and transient, so further studies on improving the capacity retention are needed.
The cycling stability of samples in high charge/discharge rate 1C are shown in Fig. 8(a). In higher charge/discharge rate, the discharge capacity of samples has all been lower than those in Fig. 7(a) as a whole, and especially in third cycles all of them all descends promptly to a stable status, obviously which is caused by faster charge/discharge speed. Even though the rapid degradation, the same tendency still exists among the materials. The samples doped with Ni exhibit better performances than pristine sample, and 5.0% Ni doped sample shows optimal capacity and retention. That means no matter in low rate or high rate, the effect of Ni element on electrochemical performance both exists, and are similar on whole tendency. We also provide the rate capability of material as shown in Fig. 8(b). Along with the increase of rate, the discharge capacity descends promptly, and when the rate goes back into 0.05C, the capacity goes up relatively, but still indicating poor rate performance of this material.
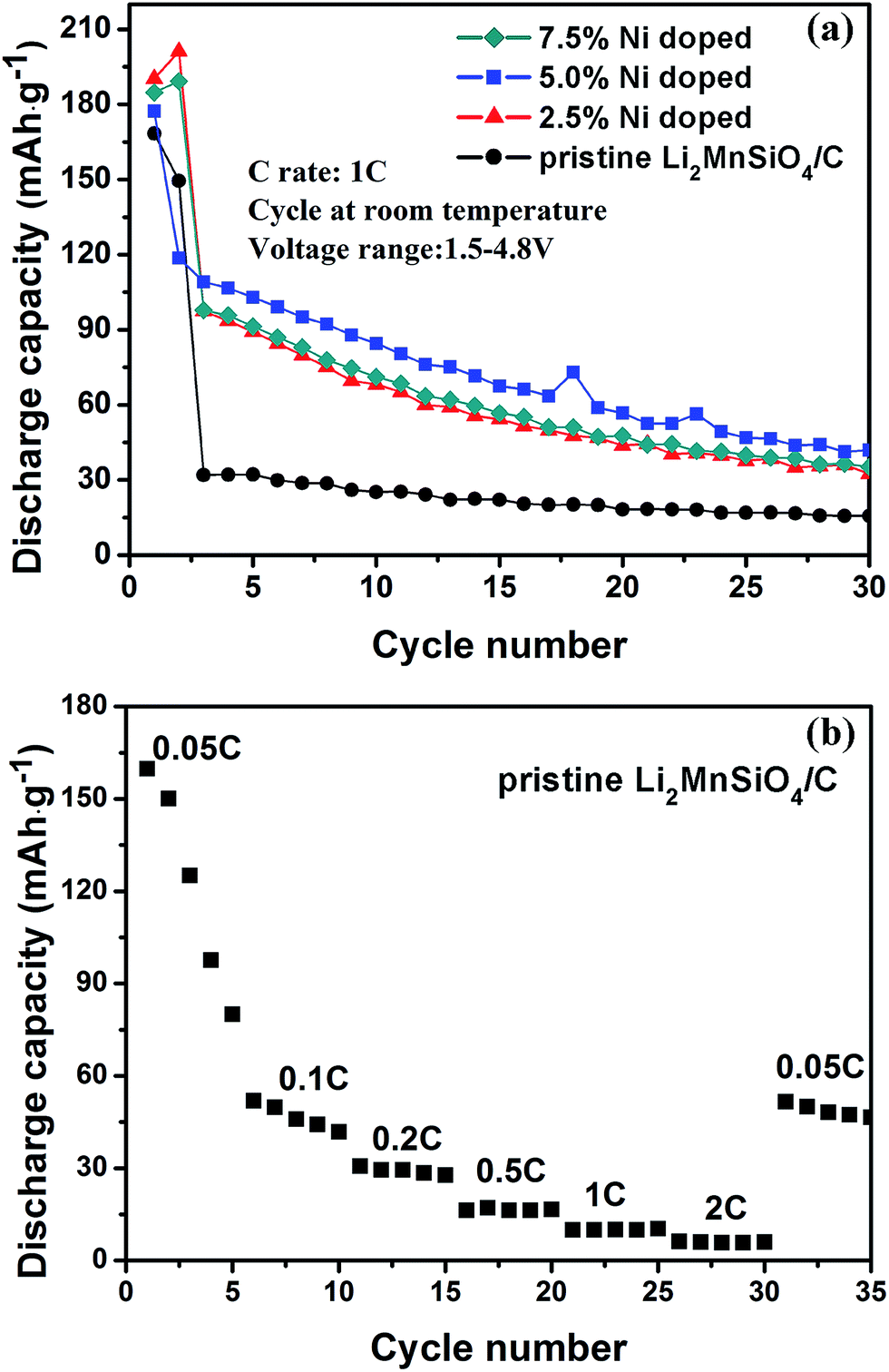 | ||
| Fig. 8 (a) The discharge capacity curves of the as-prepared Li2Mn1−xNixSiO4/C nanocomposites (cycled at room temperature, the rate is 1C, the voltage range is 1.5–4.8 V). (b) The rate capability of pristine Li2MnSiO4/C nanocomposite. | ||
To investigate the influence of Ni element substitution on the charge transfer resistance and lithium-ion diffusion coefficient, the samples are analyzed by AC impedance spectroscopy. Fig. 9(a) shows the EIS measurements of the materials at half of discharge state, and matched equivalent circuit is inserted. All the samples compose of a semi-circle in the high frequency and an inclined line in the low frequency. The semicircle can be ascribed to the combined action of charge transfer resistance (Rct) and the complex impedance of a constant phase element (CPE), while the straight line is related to the lithium-ion diffusion.45 The modified samples reveal much smaller charge transfer resistance than pristine Li2MnSiO4/C, which may behave as critical factor in promoting the electrochemical performance. Besides, among the modified samples the 5.0% Ni doped sample reveals lowest Rct value. The Rct is related to electronic conductivity and ion diffusion coefficient, so the possible explanation of the remarkable promotion is the synergistic effect of Ni and Ni2+ dopant, and there is optimal equilibrium state when 5.0% Ni doped.
 | ||
| Fig. 9 (a) EIS measurements of as-prepared Li2Mn1−xNixSiO4/C samples at half of discharge state. Matched equivalent circuit is inserted. (b) The fitting results of the latter linear part of (a). The calculated data is recorded in Table 1. | ||
The diffusion coefficient results of the lithium-ion (DLi+) can be calculated from the EIS curves of the straight line region according to the following equation.45,46

Table 2 shows the impedance results, Warburg coefficients and calculated diffusion coefficients of lithium-ion after fitting. The results display reduced charge transfer resistance and increased lithium-ion diffusion coefficient of modified samples compared to pristine sample. And the peak value also appears in 5.0% Ni doped sample, which is in good agreement with the test results above.
| x in Li2Mn1−xNixSiO4/C | Rct (Ω) | σW (Ω cm2 s−0.5) | DLi+ (cm2 s−1) |
|---|---|---|---|
| 0 | 573.0 | 162.5 | 2.94 × 10−16 |
| 2.5% | 177.3 | 86.29 | 1.05 × 10−15 |
| 5.0% | 59.88 | 30.04 | 8.64 × 10−15 |
| 7.5% | 92.92 | 48.89 | 3.26 × 10−15 |
Summing up the above discussion, the role of Ni doping is observed, and the optimal effect is all displayed for 5.0% Ni doped sample. We suggest that no matter Ni2+ or metallic Ni, they both influence the materials performance. Firstly the leading effect of Ni2+ is substituting the Mn2+ site, stabilizing the regional crystal lattice, reducing the total structural distortion extent via keeping stable ligand, and promoting voltage platform, alleviating capacity degradation. To explain the specific improvement effect of Ni2+ more visually, the models in Fig. 10 are speculated to reveal the transformation of Mn–O ligand and Ni–O ligand in oxidation reaction of pristine sample and modified samples respectively. The variation of Mn–O ligand and the Jahn–Teller effect causes structural degradation and capacity fading. The most possible oxidation process has been simulated in Fig. 10(b), where the ligand number keeps unchanged and the distortion caused by the Jahn–Teller effect is weak, so that the doped [NiO4] can sustain the whole crystal and prevent the crystal structure from collapsing so as to improve the capacity retention.
 | ||
| Fig. 10 The oxidation process of metal ligand (a) Mn–O oxide (b) Ni–O oxide. | ||
On the other hand, the metallic Ni works as conductive additive, and thus improves the poor intrinsic conductivity. However, the formation of metallic Ni would result in producing Ni vacancy, which probably leads to MnO impurity and theoretical capacity fading, meanwhile the deficiency of [NiO4] ligand would lead to regional structural distortion, thus blocking the transport of lithium-ion. In a conclusion, we think there is an optimal quantity between the two forms of Ni element to achieve best electrochemical performance, and in our work the 5.0% Ni doped sample displays optimal performance.
4. Conclusions
The concept that substitution with equivalent metal ions which can maintain unchanged ligand during oxidation/reduction process theoretically, working as structural pillars to improve electrochemical performance via alleviating the structural degradation, and then inhibiting the Jahn–Teller effect in structural variation model of Li2MnSiO4 material has been firstly hypothesized in our work. Besides, the Pmn21 samples with different Ni element content are successfully synthesized, and the existential state of Ni atom in crystalline has also been firstly discussed via XPS spectra, which proves to be coexisting metal and divalent ions.The Raman results demonstrate that transformation of [MnO4] tetrahedron to [MnO6] octahedron has been decelerated in modified samples compared with pristine Li2MnSiO4/C material, corresponding to the improved retention in electrochemical tests especially in initial several cycles. Moreover, the Ni element substitution is also effective in preparing pure phase, refining crystalline grain, promoting discharge capacity, reducing electrical resistivity and improving diffusion coefficient of lithium-ion, reflected by XRD patterns, microscope pictures and electrochemical tests.
As a conclusion, we think there are two kinds of modification mechanism with Ni element substitution at the same time. Firstly the leading effect of Ni2+ is substituting the Mn2+ site, stabilizing the regional crystal lattice, reducing the total structural distortion extent via keeping stable ligand, and thus promoting voltage platform, alleviating capacity degradation, promoting initial discharge capacity to improve electrochemical performance. The effective Ni2+ is also inferred to be limited in oxidation reactions through electrochemical results. On the other hand, although pure metallic Ni will help to reduce electrical resistivity, it still leads to formation of impurity and instability of regional structure because of deficiency of [MO4] ligand. Therefore the electrochemical performance deteriorates in high Ni element content instead. So we think there is an optimal Ni element content, under which the modified materials displays best performance. In our work, the 5.0% Ni doped sample shows optimal performance, since there is a best balance between metallic Ni and Ni2+ in 5.0% Ni doped sample.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51372136) and Shenzhen Basic Research Project (No. JCYJ20130402145002372).References
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 CAS.
- D. M. Kempaiah, D. Rangappa and I. Honma, Chem. Commun., 2012, 48, 2698–2700 RSC.
- D. Rangappa, K. D. Murukanahally, T. Tomai, A. Unemoto and I. Honma, Nano Lett., 2012, 12, 1146–1151 CrossRef CAS PubMed.
- R. J. Gummow, N. Sharma, V. K. Peterson and Y. He, J. Power Sources, 2012, 197, 231–237 CrossRef CAS.
- R. Dominko, J. Power Sources, 2008, 184, 462–468 CrossRef CAS.
- M. K. Devaraju, T. Tomai, A. Unemoto and I. Honma, RSC Adv., 2013, 3, 608–615 RSC.
- H. N. Girish and G. Q. Shao, RSC Adv., 2015, 5, 98666–98686 RSC.
- M. K. Devaraju, Q. D. Truong and I. Honma, RSC Adv., 2013, 3, 20633 RSC.
- W. Chen, M. Lan, D. Zhu, C. Wang, S. Xue, C. Yang, Z. Li, J. Zhang and L. Mi, RSC Adv., 2013, 3, 408–412 RSC.
- M. E. Arroyo-de Dompablo, M. Armand, J. M. Tarascon and U. Amador, Electrochem. Commun., 2006, 8, 1292–1298 CrossRef CAS.
- R. J. Gummow and Y. He, J. Power Sources, 2014, 253, 315–331 CrossRef CAS.
- C. A. J. Fisher, N. Kuganathan and M. S. Islam, J. Mater. Chem. A, 2013, 1, 4207 CAS.
- M. E. Arroyo-de Dompablo, R. Dominko, J. M. Gallardo-Amores, L. Dupont, G. Mali, H. Ehrenberg, J. Jamnik and E. Moran, Chem. Mater., 2008, 20, 5574–5584 CrossRef CAS.
- G. Mali, A. Meden and R. Dominko, Chem. Commun., 2010, 46, 3306–3308 RSC.
- R. Dominko, M. Bele, A. Kokalj, M. Gaberscek and J. Jamnik, J. Power Sources, 2007, 174, 457–461 CrossRef CAS.
- V. V. Politaev, A. A. Petrenko, V. B. Nalbandyan, B. S. Medvedev and E. S. Shvetsova, J. Solid State Chem., 2007, 180, 1045–1050 CrossRef CAS.
- Y.-X. Li, Z.-L. Gong and Y. Yang, J. Power Sources, 2007, 174, 528–532 CrossRef CAS.
- V. Aravindan, K. Karthikeyan, J. W. Lee, S. Madhavi and Y. S. Lee, J. Phys. D: Appl. Phys., 2011, 44, 152001 CrossRef.
- I. Belharouak, A. Abouimrane and K. Amine, J. Phys. Chem. C, 2009, 113, 20733–20737 CAS.
- Q. Zhang, Q. Zhuang, S. Xu, X. Qiu, Y. Cui, Y. Shi and Y. Qiang, Ionics, 2011, 18, 487–494 CrossRef.
- C. Lai, Z. Wang, Q. Xu and H. Li, Mater. Lett., 2015, 139, 134–137 CrossRef CAS.
- N. Kuganathan and M. S. Islam, Chem. Mater., 2009, 21, 5196–5202 CrossRef CAS.
- S. Choi, S. J. Kim, Y. J. Yun, S. S. Lee, S.-Y. Choi and H.-K. Jung, Mater. Lett., 2013, 105, 113–116 CrossRef CAS.
- M. Wang, M. Yang, L. Ma and X. Shen, RSC Adv., 2015, 5, 1612–1618 RSC.
- S. Zhang, Z. Lin, L. Ji, Y. Li, G. Xu, L. Xue, S. Li, Y. Lu, O. Toprakci and X. Zhang, J. Mater. Chem., 2012, 22, 14661 RSC.
- C. Hwang, T. Kim, Y. Noh, W. Cha, J. Shim, K. Kwak, K. M. Ok and K.-K. Lee, Mater. Lett., 2016, 164, 270–273 CrossRef CAS.
- M. Zhang, S. Zhao, Q. Chen and G. Yan, RSC Adv., 2014, 4, 30876 RSC.
- R. Chen, R. Heinzmann, S. Mangold, V. S. K. Chakravadhanula, H. Hahn and S. Indris, J. Phys. Chem. C, 2013, 117, 884–893 CAS.
- S. Zhang, Y. Li, G. Xu, S. Li, Y. Lu, O. Toprakci and X. Zhang, J. Power Sources, 2012, 213, 10–15 CrossRef CAS.
- M. Bini, S. Ferrari, D. Capsoni, C. Spreafico, C. Tealdi and P. Mustarelli, J. Solid State Chem., 2013, 200, 70–75 CrossRef CAS.
- R. Dominko, I. Arčon, A. Kodre, D. Hanžel and M. Gaberšček, J. Power Sources, 2009, 189, 51–58 CrossRef CAS.
- A. Saracibar, Z. Wang, K. J. Carroll, Y. S. Meng and M. E. A.-d. Dompablo, J. Mater. Chem. A, 2015, 3, 6004–6011 CAS.
- Y.-C. Wang, S.-X. Zhao, P.-Y. Zhai, F. Li and C.-W. Nan, J. Alloys Compd., 2014, 614, 271–276 CrossRef CAS.
- R. Dominko, M. Bele, M. Gaberšček, A. Meden, M. Remškar and J. Jamnik, Electrochem. Commun., 2006, 8, 217–222 CrossRef CAS.
- Y. Zhao, C. Wu, J. Li and L. Guan, J. Mater. Chem. A, 2013, 1, 3856 CAS.
- H. Gong, Y. Zhu, L. Wang, D. Wei, J. Liang and Y. Qian, J. Power Sources, 2014, 246, 192–197 CrossRef CAS.
- Q. Liu, W. Liu, D. Li and Z. Wang, Mater. Lett., 2016, 162, 87–90 CrossRef CAS.
- T. Muraliganth, K. R. Stroukoff and A. Manthiram, Chem. Mater., 2010, 22, 5754–5761 CrossRef CAS.
- M. Kim, X. M. Chen, X. Wang, C. S. Nelson, R. Budakian, P. Abbamonte and S. L. Cooper, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 174424 CrossRef.
- P.-Y. Zhai, S.-X. Zhao, H.-M. Cheng, J.-W. Zhao and C.-W. Nan, Electrochim. Acta, 2015, 153, 217–224 CrossRef CAS.
- C. M. Julien and M. Massot, Mater. Sci. Eng., B, 2003, 97, 217–230 CrossRef.
- S. H. Oh, K. Y. Chung, S. H. Jeon, C. S. Kim, W. I. Cho and B. W. Cho, J. Alloys Compd., 2009, 469, 244–250 CrossRef CAS.
- G. B. Zhong, Y. Y. Wang, X. J. Zhao, Q. S. Wang, Y. Yu and C. H. Chen, J. Power Sources, 2012, 216, 368–375 CrossRef CAS.
- Y. Xue, Z. Wang, F. Yu, Y. Zhang and G. Yin, J. Mater. Chem. A, 2014, 2, 4185 CAS.
- J. Zhu, Z. Tang, H. Tang, Q. Xu and X. Zhang, J. Electroanal. Chem., 2016, 761, 37–45 CrossRef CAS.
- J. Zhang, J. Shen, T. Wang, C. Wei, Y. Ma, C. Zhu and Y. Yue, Electrochim. Acta, 2013, 111, 691–697 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |