
A new POM–MOF hybrid microporous material with ultrahigh thermal stability and selective adsorption of organic dyes†
Miao Huoab,
Wenbin Yang*a,
Hailong Zhangab,
Lei Zhanga,
Jianzhen Liaoab,
Lang Lina and
Canzhong Lu*a
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China. E-mail: czlu@fjirsm.ac.cn; Fax: +86-591-83714946; Tel: +86-591-83705794
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 16th November 2016
Abstract
Using Keggin heteropolyoxometalate H3PW12O40 as the template, a new host–guest POM–MOF hybrid material formulated as (CH3NH2CH3)[Cu2(TPB)2 (PW12O40)]·4DMF·6H2O (1) has been successfully synthesized under solvothermal conditions. Complex 1 is characteristic of a 3D host framework constructed from Cu+ ions and TPB ligands and containing different types of one-dimensional channels. The larger channels along the c axis are occupied by Keggin polyoxoanions as guests, while the narrower ones along the [110] direction are available to accommodate solvent molecules and counter dimethylamine cations. Complex 1 shows ultrahigh thermal stability and permanent porosity. In addition, dye adsorption and photocatalytic properties of 1 have been investigated. The results indicated that complex 1 is not only a good heterogeneous photocatalyst for the degradation of MO−, but also selectively captures MB+ from binary mixtures of MB+/MO−, MB+/R6G+ or MB+/RhB+.
Introduction
Polyoxometalates (POMs) are a kind of anionic inorganic metal–oxygen cluster of early transition metal elements in high oxidation states (such as Mo6+, V5+, W6+ and Nb5+), and have abundant topologies and compositions, acid/base tenability, diverse electronic and photochemical properties, oxygen-rich surface and controllable sizes.1 Although POMs were discovered more than two centuries ago and a huge number of polyoxometalates have been reported so far, new research hotspots on POMs still pop up from time to time.2–4 For example, the use of POMs as inorganic building blocks to construct inorganic–organic hybrid materials with various metal–organic coordination fragments has become a new research topic in recent material science.2,4,5 In these hybrids, the polyoxometalate anions are either coordinated to secondary metal atoms via metal–oxygen coordination bonds, or connected as guests/templates/counter ions through non-covalent interactions.2–5 As a result, the POM-based hybrid materials combine the advantages of inorganic polyoxometalates and the functionalities of organic components, which make them more attractive for many new potential applications in catalysis, medicine, magnetism, gas storage, chemical separation, ion exchange reactions and optics.Due to their high surface areas, meaningful physicochemical stability and adjustable pore structures, metal–organic frameworks (MOFs) have been applied widely in various areas6–13 and play an eventful platform in host–guest chemistry of polyoxometalates (POMs). MOFs with nano cages or channels can provide unique chemical environments for accommodating POMs to form host–guest hybrid materials with interesting properties.14 To obtain POM–MOF host–guest hybrids, the guest of POMs can either be hosted in the pre-synthesized MOFs with suitable nano pores15–17 or directly assembled with metal cations and organic ligands in one-pot synthesis reactions.18 In the process of assembling POM–MOF host–guest hybrids, POMs can not only induce metal cations together with organic ligands to form nanoporous MOFs when working as templates, but also fill in the gaps of MOFs to effectively avoid the formation of interpenetrating structures. In addition, POMs usually have a high negative charge, balancing the charge and stabilizing the structure of the host frameworks. In principle, the structure of POM–MOF host–guest composites can be regulated by choosing different types of POMs19 among which the Keggin heteropolyoxometalates have received particular attention due to their commercial availability or easy preparation, nanoscale dimensions, and good coordination abilities. In the past years, our research group has developed a series of POM-based metal–organic pseudorotaxane frameworks, POM-based supramolecular aggregates and POM–MOF host–guest hybrids using Keggin-type POMs as building blocks in the presence of polydentate N-donor ligands.15,20–22
Another important factor in the synthesis of POM–MOF hybrids is to choose a suitable organic ligand because the length, geometry, and coordination ability of the ligands all play significant roles in generating frameworks.23 Herein, we choose 1,2,4,5-tetra(4-pyridyl)benzene (TPB) as a new rigid multifunctional ligand, which can provide with four nitrogen atoms available to coordinate. The TPB ligand is a kind of tetra-dentate nitrogen-containing ligands with novel structural features which can make it possible to realize host frameworks with specific topologies.24,25 Conformational freedom of the TPB ligand through rotation of the C–C bonds between pyridine and central benzene rings causes the variation in structural topology, which imparts them not only intriguingly different luminescence properties but also different morphologies when co-crystallized with different solvents.25–27 More recently, a great progress has been made in obtaining POM–MOF host–guest hybrids through using TPB ligand, and herein, we report a POM-templated host–guest compound, (CH3NH2CH3)[Cu2(TPB)2 (PW12O40)]·4DMF·6H2O (1, DMF = N,N-dimethylformamide). In 1, the nanosized Keggin anion [PW12O40]3− was incorporated as guests into one kind of 1D channels of the host framework. In addition, the photocatalytic properties, selective adsorption ability toward organic dyes and gas adsorption of 1 have been investigated.
Experimental section
Materials and general methods
The TPB ligand was synthesized according to the reported literature method.26 The other reagents and solvents for synthesis were purchased from commercial sources, and used without further purification. Elemental analysis of C, H, and N were performed on an Elementar Vario EL III microanalyzer. The IR spectra were obtained on a Vertex 70 Spectrum with KBr pellet in the 400–4000 cm−1 region. Thermal gravimetric analysis studies were performed with a TGA/DSC 1 STARe system at a rate of 10 °C min−1 under N2 atmosphere. Powder X-ray diffraction data were recorded on a Mini FlexII powder diffractometer with graphite monochromatized Cu-Kα radiation (λ = 1.54056 Å). Optical diffuse reflectance spectra were measured on PEL Ambda 900 UV-vis spectrophotometer using BaSO4 plate as the reference. Photocatalysis of compound 1 was carried out with a 300 W Xe lamp as a visible light source and analyzed by UV-visible spectroscopy. Single component gas adsorption measurements were collected in the Accelerated Surface Area and Porosimetry 2020 System (ASAP2020).Synthesis of 1
A mixture of CuCl (0.050 g, 0.500 mmol), TPB (0.01 g, 0.026 mmol), and H3PW12O40·xH2O (0.150 g, 0.052 mmol) was dissolved in 5 mL DMF and 5 mL acetonitrile at room temperature. After stirring for 30 min at room temperature, the reaction suspension was transferred and sealed in a Parr Teflon-lined stainless steel vessel (20 mL), kept at 160 °C under autogenous pressure for two days, and then cooled slowly to room temperature over two days, resulting in brown crystals of 1. The crystals were collected by filtration, washed with H2O, and then dried in open air (about 36% yield based on copper). Anal. calcd (found%) for 1: C18.75 (18.19), H 1.98 (1.93), N 4.31 (4.21). IR (KBr, cm−1): 3440(s), 1610(s), 1544(w), 1479(w), 1421(m), 1382(w), 1218(w), 1076(s), 978(s), 893 (s), 815(s), 594(w), 554(w).X-ray crystallography
Single-crystal X-ray diffraction data of complex 1 were recorded on an Agilent Technologies Super-Nova diffractometer equipped with Atlas CCD detector using monochromated Cu Kα radiation (λ = 1.54184 Å) at 293 K. The data were corrected for absorption using multi-scan technique as implemented in SADABS. The structures were solved by direct methods and refined by full-matrix least-squares on F2 using the SHELXTL software package.28,29 All non-hydrogen atoms of polyoxometalate anion and Cu–organic framework are refined with anisotropic parameters, and all H atoms on carbon atoms were generated geometrically and refined in the riding model using isotropic displacement parameters. For 1, the solvent molecules were highly disordered, and could not be modeled properly, thus, a PLATON/SQUEEZE procedure30 was employed to calculate the diffraction contribution of solvent molecules and, thereby, to produce a set of solvent-free diffraction intensities. The final formula of 1 was determined from the SQUEEZE results combined with elemental analysis and TGA data, showing that there are approximately 4 DMF, 6H2O and one (CH3)2NH2+ cation per formula. The details for data collection and structural refinements are summarized in Table 1, and selected bond lengths and angles are given in the ESI.† The phase purity of as-synthesized 1 is confirmed by the PXRD measurement (ESI†). The CCDC (1473396) is for 1.| a R1 = ∑||Fo| − |Fc||/∑|Fo|.b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. | |
|---|---|
| Compound | 1 |
| Empirical formula | C66H84Cu2N13O50PW12 |
| Formula weight | 4223.23 |
| Crystal system | Tetragonal |
| Space group | I41/acd |
| a/Å | 26.9629(3) |
| b/Å | 26.9629(3) |
| c/Å | 27.5365(4) |
| α/° | 90.00 |
| β/° | 90.00 |
| γ/° | 90.00 |
| Volume/Å3 | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 018.9(4) 018.9(4) |
| Z | 8 |
| ρcalc/g cm−3 | 2.803 |
| μ/mm−1 | 26.13 |
| F(000) | 15456.0 |
| Reflections collected | 34445 |
| Unique reflections | 4809 (Rint = 0.0371) |
| Restraints/parameters | 3/273 |
| Goodness-of-fit on F2 | 1.144 |
| Final R indexes [I ≥ 2σ (I)] | aR1 = 0.0407, bwR2 = 0.1065 |
| Final R indexes [all data] | R1 = 0.0480, wR2 = 0.1146 |
| Largest diff. peak/hole/e Å−3 | 1.402/−1.885 |
Dye molecule adsorption experiments for 1
Adsorption experiments including adsorption equilibrium and selective adsorption experiments have been investigated. Fresh as-synthesized crystalline samples of 1 were immersed into the dye solution at room temperature in a beaker by simultaneous stirring. At a given time interval, the concentrations of dyes were monitored with UV-visible spectra at the maximum absorbance of each dye (664 nm, 464 nm, 527 nm, 554 nm for MB+, MO− and R6G+, RhB+, respectively). The details of each type of experiments are given in the ESI.†MO photodegradation using 1
As-synthesized crystalline sample of 1 (50.0 mg) and hydrogen peroxide (30%, 2.0 mL) were dispersed in an aqueous solution of methyl orange (MO−, 10 mg L−1, 100 mL) under stirring, and then irradiated with a 300 W Xe lamp as a UV-visible light source. A small amount of the mixed solution were filtered from the reactor intermittently during the illumination, and evaluated by measuring characteristic absorption peak of MO− solution after a regular interval visible light irradiation. For comparison, blank experiments in the absence of 1 and in the dark were also performed.Structure description
Single-crystal X-ray diffraction analysis revealed that complex 1 crystallizes in the tetragonal I41/acd space group, and its structure is constructed from a 3D non-interpenetrating copper–organic framework with nano channels occupied by Keggin anions. There is only one crystallographically independent Cu atom (Fig. 1a) in the asymmetric unit of 1, and the Cu atom resides in a tetrahedral coordination geometry defined by four N atoms from four different TPB ligands. All Cu–N bond lengths (1.999(7)–2.033(7) Å) and N–Cu–N bond angles (105.3(4)–115.9(4)°) are in the normal range reported for tetrahedral Cu(I) ion. The TPB ligand exhibits only one coordination mode, and links four different Cu(I) atoms via four outer pyridines (Fig. 1a). The interconnection of Cu(I) ions and TPB ligands affords a 3D metal–organic framework with different types of one-dimensional channels. As shown in Fig. 1b and c, the half of TPB ligands (namely, cis-position pyridyl moieties in TPB ligands) link tetrahedral Cu(I) ions to construct a small single-stranded helix running along a 41 screw axis, and the screw pitch is equal to the unit cell parameter c [27.5365(4) Å]. Because of the geometry of TPB ligands (a symmetric center located in the central benzene of TPB ligand) and the symmetrical operations of the space group of 1, the small helix (or helical channels A) are further aligned side by side and interconnected by sharing TPB ligands to form a 3D framework, and afford another type of larger channels (channels B) of diameter 1.2 nm. As shown in Fig. 1c, channels B are surrounded by the aromatic faces of phenyl groups and coordinated pyridine rings of TPB ligands, and are filled by [PW12O40]3− polyoxoanion (Fig. 1c).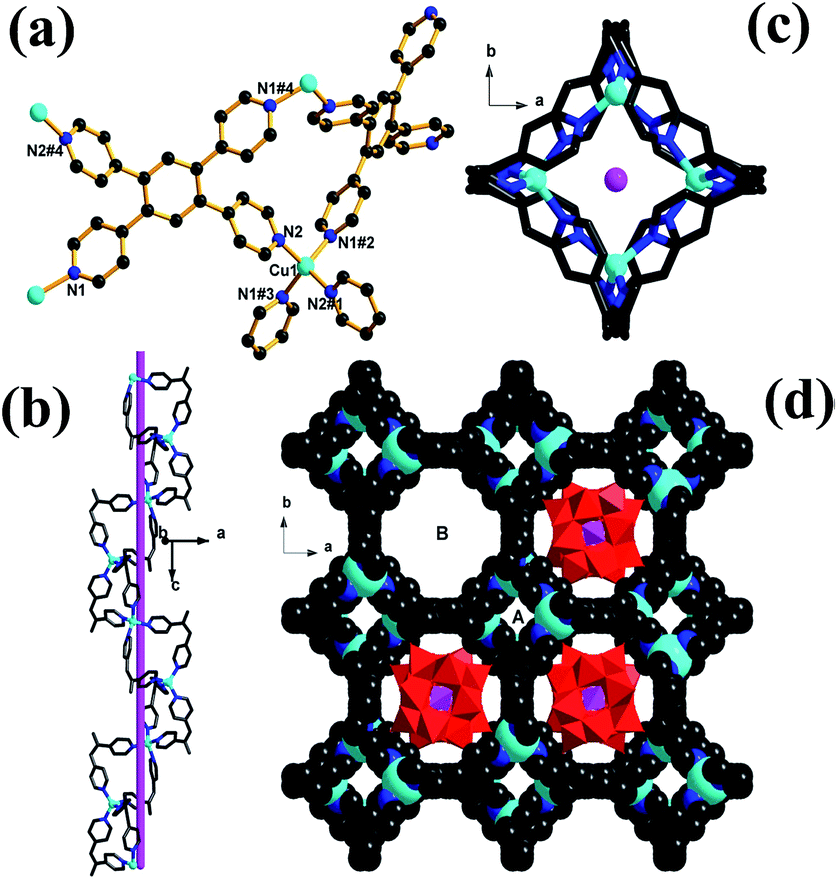 | ||
| Fig. 1 (a) The coordination environments of Cu(I) and the coordination mode of TPB in 1. (b) and (c) a view of the small helical tubes (or helical channels A), which are interconnected into a 3D framework with another type of larger helical channels B occupied by Keggin polyoxoanions (d). Keggin anions highlighted in polyhedra, and color code: C, dark grey; N, blue; Cu, turquoise. | ||
To illustrate the unique structure of 1, each Cu centre, which is connected to four TPB ligands, can be represented as a four-connecting tetrahedral node, while the TPB ligand can be depicted as a 4-connecting rectangular node (Fig. 2a). Thus, the host framework structure of 1 is simplified into a sqc net (Fig. 2b) with the Schläfli symbol of (42·83·10)(42·84). From the tiling aspect (Fig. 2c), the larger channels B in the host framework possess nanosize pores which are filled with Keggin anions, while channels A is too small to accommodate any solvent molecules. However, when viewed along the [110] direction, there are the third kind of one-dimension channels in diameter of ca. 0.5 nm (Fig. S2†), which remain void and are available to capture guest solvent molecules and counter cations (CH3)2NH2+.
 | ||
| Fig. 2 Topological network of the host framework in 1. (a) Each TPB ligand can be described as a 4-connecting rectangular node. (b) a polyhedral view of the (4,4)-connected sqcnet by depicting the TPB ligand as a 4-connecting rectangle (blue) and the metal as a 4-connecting tetrahedral node (purple). The black lines are the linkage between these two kinds of nodes. (c) The host framework illustrated as a natural tiling. | ||
Thermal stability and N2 sorption of 1
In order to test the permanent porosity of 1, N2 adsorption experiments were carried out. Remarkably, even though the large channels of the host framework are filled by the nanosize Keggin polyoxoanions, the theoretical solvent-accessible free volume of 1 was still estimated by PLATON30 to be 34.8% of the total unit cell volume. The thermogravimetric analysis (TGA) analysis of 1, performed under N2 atmosphere, revealed a weight loss of 2.4% (calcd 2.6%) from 30 °C to 100 °C, corresponding to the release of guest water molecules, and a weight loss of 7.5% (calcd 7.2%) from 120 °C to 340 °C, corresponding to the release of trapped guest DMF molecules and dimethylamine cations. The resulting solid shows a considerable steady plateau until 590 °C beyond which the framework starts to decompose (Fig. S4, ESI†). The fully desolvated framework, obtained by heating the methanol-exchanged sample of 1 at 100 °C under dynamic vacuum for 10 hours, remains crystalline with the observed PXRD pattern matching well the calculated pattern derived from the single crystal structure of 1 (Fig. S5†). As shown in Fig. S6,† upon thermal treatments at different temperatures, the PXRD patterns of 1 remain almost intact up to 550 °C although some diffraction peaks become slightly weaker, confirming the maintenance of framework integrity. Such ultrahigh thermal stability has never been reported for POM-based hybrid materials.15–20 The permanent porosity of 1 was supported by the reversible type-I N2 adsorption isotherm (Fig. 3) at 77 K characteristic of a microporous material. Based on the N2 adsorption isotherm at 77 K, the Brunauer–Emmett–Teller (BET) and Langmuir surface areas were estimated to be 257.7 and 361.6 m2 g−1, respectively, and the pore volume is calculated to be 0.129 cm3 g−1. Meanwhile, the pore sizes calculated from analysis of the N2 isotherm at 77 K using the Horvath–Kawazoe (H–K) model are distributed around 0.49 nm, consistent well with the channel diameters estimated from the single-crystal structure determination.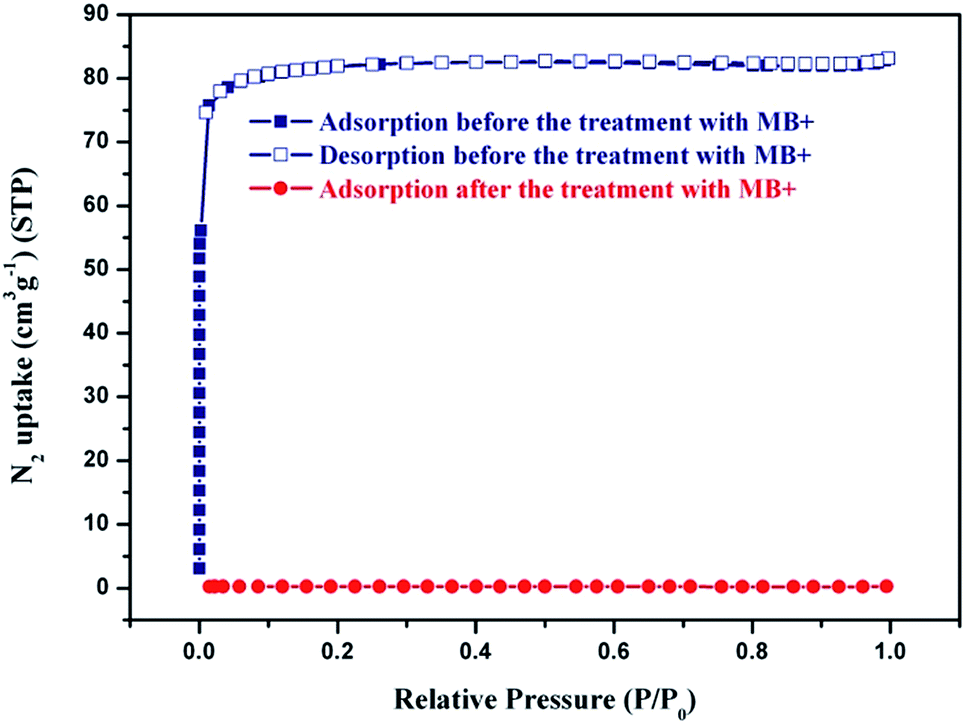 | ||
| Fig. 3 N2 adsorption isotherms of 1 at 77 K before and after the treatments with MB+. | ||
Dye molecule selective adsorption on 1
Organic dyes play an important role in human life, and using POMs and related composites to photocatalytically degrade organic dye contamination has drawn the attention of modern researchers in recent years. However, some composites show poor degradability due to their structural limits. Thus, from the recycling point of view, it is more attractive to capture and separate different organic dyes by using the capture ability of POMs composites. When 1 was added to an aqueous solution of MB+ and stirred for a while, the colour of the crystalline sample of 1 changed rapidly from brown to dark green while the colour of MB+ solution varied from deep blue to very pale blue, and the uptake of MB+ on 1 is almost linear to the staring concentration below 50 mg L−1 (Fig. S9†), suggesting that 1 has significant adsorption ability of MB+ molecules. To fully evaluate the adsorption properties of 1, the same amount of crystalline sample of 1 was dispensed into an aqueous solution with the same volume and concentration of methylene blue (MB+), rhodamine 6G (R6G+) and methyl orange (MO−) under stirring in dark, and at given time intervals, the concentration change of dye solution was monitored by the UV-vis adsorption spectra. These dye molecules have different sizes and charges: the positively charged MB+ and R6G+ with different molecule sizes; the opposite charged MB+ and MO− with the similar molecule size. As shown in Fig. 4, the experimental results revealed that the adsorption of MO− and R6G+ on 1 was very small, in contrast to the strong adsorption towards MB+. Interestingly, the N2 adsorption isotherms revealed that the uptakes of N2 at 77 K after the treatment with MB+ are almost negligible (Fig. 3), for example, the uptake of N2 at P/P0 = 0.9 is only 0.24 cm3 g−1 significantly lower than the value (81.9 cm3 g−1) before the treatment with MB+, indicating that MB really enters the pores of the material, and the pores of the material are blocked completely by MB+ molecules. | ||
| Fig. 4 The adsorption activity of 1 toward different dye aqueous solutions. | ||
The adsorption selectivity of 1 is further investigated by applying a binary mixture of MB+/R6G+ (positively charged with different molecular sizes) or MB+/MO− (similar sizes with different charge). From the Fig. 5, it can be seen that complex 1 can selectively capture MB+ molecules from the corresponding binary mixture, along with the UV-vis adsorption characteristic peak of MB+ almost disappeared, and leaving the characteristic peak of R6G+, RhB+ or MO−. After two hours, complex 1 absorbs more than 98% MB+ from the mixed dye solution. In comparison, only 18.06% R6G+, 14.3% RhB+ or 3.04% MO− is absorbed. These studies suggest that the charge and size of organic dyes are the two significant factors to be considered for capture and separate organic dyes using POM composites.
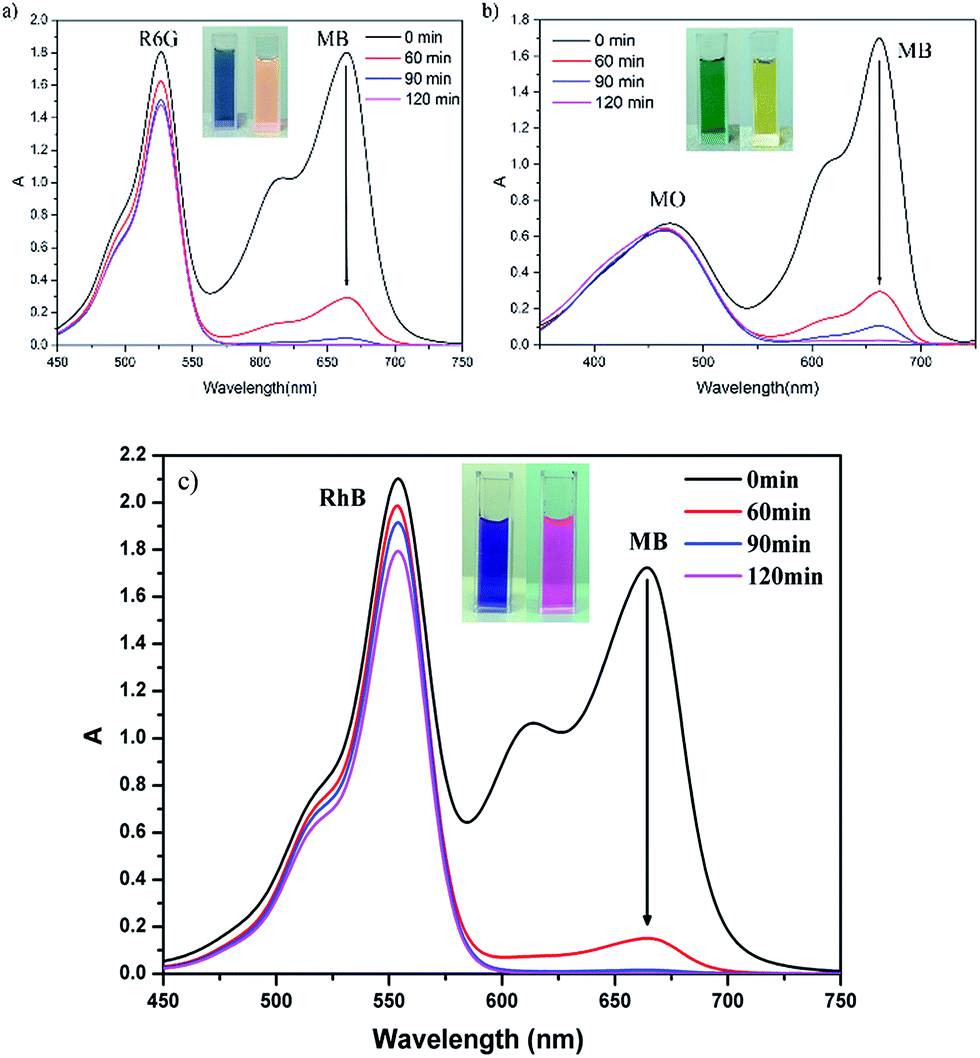 | ||
| Fig. 5 (a) The selective adsorption capability of 1 toward the mixed solution of R6G+ and MB+. (b) The selective adsorption capability of 1 toward the mixed solution of MO− and MB+. (c) The selective adsorption capability of 1 toward the mixed solution of RhB+ and MB+. | ||
To study the release ability of MB+ adsorbed, crystalline sample of 1 was immersed in an aqueous solution (20 mg L−1, 100 mL) of MB+ under stirring for three days. Upon loading of MB+ in 1, the deep blue powder solid MB+@1 obtained by filtration and washing quickly with deionized water was immersed into a pure water (100 mL) under stirring. The release process was monitored and calculated by liquid phase UV-visible spectra. Interestingly, after stirring for 6 hours, the desorption amount of MB+ is still close to zero, indicating that the adsorbed MB+ molecules have very strong interactions with 1. The desorption of MB+ on 1 is thus very difficult, which is a downside from the point of recovering absorbent, however it makes the adsorption more thoroughly.
Obviously, the adsorption selectivity of 1 towards organic dyes is not only related to the structure of 1 but also affected by the charge and molecular size of organic dyes. Firstly, the host framework of 1 is filled with polyoxometalate anions and therefore negatively charged. Positively charged MB+ can easily combine with the active adsorption sites of 1 through electrostatic interactions, and complex 1 therefore demonstrates good adsorption ability toward MB+. Due to the strong forces between small cationic MB+ molecules and the host framework of 1, the absorbed MB+ molecules become difficult to be released. As for cationic R6G+, the larger molecular volume will result in a steric hindrance when binding to the active adsorption sites on 1, and thus shows a significantly lower adsorption rate. Although MO− molecules has similar molecule size with MB+, a very low uptake capacity was observed for MO− as negative charge of this dye molecule which causes charge repulsion between MO− molecules and active adsorption sites on 1.
Photocatalytic properties of 1
Although organic dyes are very useful in industrial and agricultural settings, it is difficult to degrade organic dyes in waste water. Previous reports31–33 have showed that POMs and POMs-based hybrid materials are efficient photocatalysts on degradation of organic dyes. Because of its microporous features, the presence of Keggin anions, and ultrahigh stability, complex 1 was chosen as a catalyst candidate to investigate the photodegradation of methyl orange (MO−), which is a common pollution source in environments. 50 mg of complex 1 was dispersed in 100 mL of MO− solution (10 mg L−1) containing 2 mL H2O2 (30%), and the reaction mixture was stirred and irradiated with 300 W Xe lamp as the visible light source. After every 15 minute time interval, 3 mL of solution was collected by filtration and used to monitor the degradation of MO− through testing the characteristic absorption peak of MO− at 464 nm with UV-vis spectra (Fig. 6a). Control experiments were also carried out either in dark or without 1 (Fig. 6b). As shown in Fig. 6a, the characteristic adsorption peaks of MO solution are decreased obviously with the increasing of reaction time. Under visible light irradiation and in the presence of 1, the degradation of MO− can reach up to 98.8% within two hours. In comparison, the characteristic adsorption peaks of MO− solution in the dark or in the absence of 1 show no obvious change and the final decolourizations of MO− are less than 10% (in dark) and 16.5% (without 1), respectively. In addition, the photocatalytic degradation remarkably decreases with the addition of IPA (a quencher of ˙OH). Therefore, it can be concluded that the ˙OH species play a major role in the photocatalytic degradation of MO, and the photocatalytic degradation mechanism of MO over 1 may be proposed in Scheme S1.† The copper(I)–organic framework ([Cu2(TPB)2]2+) acts as sensitizer (S) to be excited by UV-vis light, and the photo-generated electrons transfer to the LUMO of Keggin POM ([PW12O40]3−). The adsorbed H2O2 (major) and O2 (minor) trap an electron in LUMO of the POM to generate oxidant radicals ˙OH and O2˙−. Some of the O2˙− radicals further react with H2O2 to produce ˙OH.34 Then, the ˙OH radicals, together with O2˙−, attack and oxidize organic dye molecules (MO). Similar photo-catalytic degradation processes were observed in POM-based complexes.34–37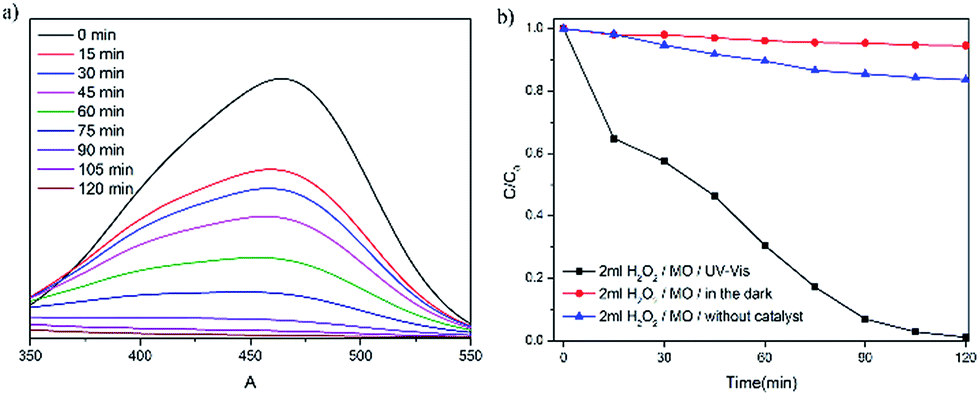 | ||
| Fig. 6 (a) UV-vis absorption spectra of the MO− solution during the decomposition reaction in the presence of 1 under visible light irradiation. (b) Changes in C/C0 plots of MO− versus reaction time (1) in the presence of 1 and under visible light irradiation (2) in the dark and (3) without catalyst. | ||
The PXRD patterns of the powder solid recycled after degradation of MO− are well consistent with the simulated one from single-crystal structure, suggesting that the framework structure of 1 remain undamaged. Furthermore, successive photocatalytic experiments performed with the powder recycled from the reaction suspension by simple filtration revealed that the activity of the recovered catalyst does not significantly decrease. These above results denoted that complex 1 has the potential to be a good heterogeneous photocatalyst on the degradation of MO− organic dye.
Conclusions
In summary, we have successfully synthesized a new interesting host–guest POM–MOF hybrid material, with the formula of (CH3NH2CH3)[Cu2(TPB)2(PW12O40)]·4 DMF·6H2O (1). Single-crystal diffraction analysis revealed that complex 1 is characteristic of a 3D host framework constructed from Cu+ ions and TPB ligands and containing different types of one-dimension channels. The larger channels along the c axis are occupied by Keggin polyoxoanions as guests, while the narrower ones along the [110] direction stay are available to accommodate solvent molecules and counter dimethylamine cations. In this work, templates play an important role in the structural control of 1. In addition, dye adsorption, photocatalytic properties of 1 have been investigated. The results indicated that complex 1 is not only a good heterogeneous photocatalyst on the degradation of MO− in the presence of H2O2, but also selectively captures MB+ from binary mixtures of MB+/MO−, MB+/R6G+ or MB+/RhB+.Acknowledgements
This work was supported by the 973 key program of the Chinese Ministry of Science and Technology (MOST) (2012CB821705), the Chinese Academy of Sciences (KJCX2-YW-319, KJCX2-EW-H01), the National Natural Science Foundation of China (21373221, 21521061, 91122027, 51172232, 21403236) and the Natural Science Foundation of Fujian Province (2012J06006, 2014J05026, 2006L2005).Notes and references
- M. Nyman and P. C. Burns, Chem. Soc. Rev., 2012, 41, 7354–7367 RSC.
- D. L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC.
- E. D. Anne Dolbecq, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef PubMed.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 RSC.
- E. Cadot, M. N. Sokolov, V. P. Fedin, C. Simonnet-Jegat, S. Floquet and F. Secheresse, Chem. Soc. Rev., 2012, 41, 7335–7353 RSC.
- S.-T. Zheng, J. T. Bu, Y. Li, T. Wu, F. Zuo, P. Feng and X. Bu, J. Am. Chem. Soc., 2010, 132, 17062–17064 CrossRef CAS PubMed.
- Q. Lin, T. Wu, S. T. Zheng, X. Bu and P. Feng, J. Am. Chem. Soc., 2012, 134, 784–787 CrossRef CAS PubMed.
- P.-Q. Liao, D.-D. Zhou, A.-X. Zhu, L. Jiang, R.-B. Lin, J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2012, 134, 17380–17383 CrossRef CAS PubMed.
- S. C. Xiang, Z. Zhang, C. G. Zhao, K. Hong, X. Zhao, D. R. Ding, M. H. Xie, C. D. Wu, M. C. Das, R. Gill, K. M. Thomas and B. Chen, Nat. Commun., 2011, 2, 204–210 CrossRef PubMed.
- S. S. Chen, P. Wang, S. Takamizawa, T. A. Okamura, M. Chen and W. Y. Sun, Dalton Trans., 2014, 43, 6012–6020 RSC.
- S. Yang, S. K. Callear, A. J. Ramirez-Cuesta, W. I. F. David, J. Sun, A. J. Blake, N. R. Champness and M. Schröder, Faraday Discuss., 2011, 151, 19 RSC.
- S.-Y. Liu, X.-L. Qi, R.-B. Lin, X.-N. Cheng, P.-Q. Liao, J.-P. Zhang and X.-M. Chen, Adv. Funct. Mater., 2014, 24, 5866–5872 CrossRef CAS.
- W. Zhu, C. He, P. Wu, X. Wu and C. Duan, Dalton Trans., 2012, 41, 3072–3077 RSC.
- D. Y. Du, J. S. Qin, S. L. Li, Z. M. Su and Y. Q. Lan, Chem. Soc. Rev., 2014, 43, 4615–4632 RSC.
- X. Kuang, X. Wu, R. Yu, J. P. Donahue, J. Huang and C. Z. Lu, Nat. Chem., 2010, 2, 461–465 CrossRef CAS PubMed.
- F. J. Ma, S. X. Liu, C. Y. Sun, D. D. Liang, G. J. Ren, F. Wei, Y. G. Chen and Z. M. Su, J. Am. Chem. Soc., 2011, 133, 4178–4181 CrossRef CAS PubMed.
- C. Duan, M. Wei, D. Guo, C. He and Q. Meng, J. Am. Chem. Soc., 2010, 132, 3321–3330 CrossRef CAS PubMed.
- S. Freye, R. Michel, D. Stalke, M. Pawliczek, H. Frauendorf and G. H. Clever, J. Am. Chem. Soc., 2013, 135, 8476–8479 CrossRef CAS PubMed.
- F.-J. Ma, S.-x. Liu, C.-y. Sun, D.-D. Liang, G.-j. Ren, F. Wei, Y.-g. Chen and Z.-m. Su, J. Am. Chem. Soc., 2011, 133, 4178–4181 CrossRef CAS PubMed.
- X. Kuang, X. Y. Wu, J. Zhang and C. Z. Lu, Chem. Commun., 2011, 47, 4150–4152 RSC.
- L. Zhang, W. Yang, X. Kuang, X. Wu and C. Lu, Dalton Trans., 2014, 43, 16328–16334 RSC.
- X.-J. Dui, W.-B. Yang, X.-Y. Wu, X. Kuang, J.-Z. Liao, R. Yu and C.-Z. Lu, Dalton Trans., 2015, 44, 9496–9505 RSC.
- H. Zhao, Z.-R. Qu, H.-Y. Ye and R.-G. Xiong, Chem. Soc. Rev., 2008, 37, 84–100 RSC.
- N. Zheng, X. Bu and P. Feng, J. Am. Chem. Soc., 2002, 124, 9688–9689 CrossRef CAS PubMed.
- S. Hu, Z.-S. Meng and M.-L. Tong, Cryst. Growth Des., 2010, 10, 1742–1748 CAS.
- Y. C. Chang and S. L. Wang, J. Am. Chem. Soc., 2012, 134, 9848–9851 CrossRef CAS PubMed.
- L. Han, W. Zhao, Y. Zhou, X. Li and J. Pan, Cryst. Growth Des., 2008, 8, 3504–3507 CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- J. Lu, J. X. Lin, X. L. Zhao and R. Cao, Chem. Commun., 2012, 48, 669–671 RSC.
- Y. Hu, F. Luo and F. Dong, Chem. Commun., 2011, 47, 761–763 RSC.
- H. Lin and P. A. Maggard, Inorg. Chem., 2008, 47, 8044–8052 CrossRef CAS PubMed.
- L. Zhang, B. Shan, H. Yang, D. Wu, R. Zhu, J. Niu and R. Cao, RSC Adv., 2015, 5, 23556–23562 RSC.
- X. L. Wang, C. H. Gong, J. W. Zhang, G. C. Liu, X. M. Kan and X. Nu, CrystEngComm, 2015, 17, 4179–4189 RSC.
- W. M. Xiao, P. P. Zhao, S. J. Deng and N. Zhang, New J. Chem., 2015, 39, 3719–3727 RSC.
- X. J. Dui, W. B. Yang, X. Y. Wu, X. F. Kuang, J. Z. Liao, R. M. Yu and C. Z. Lu, Dalton Trans., 2015, 44, 9496–9505 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Selected bond lengths and angles, additional images, IR, TGA, PXRD, EDS data. CCDC 1473396. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra10422c |
| This journal is © The Royal Society of Chemistry 2016 |