
Anti selective glycolate aldol reactions of (S)-4-isopropyl-1-[(R)-1-phenylethyl]imidazolidin-2-one: application towards the asymmetric synthesis of 8-4′-oxyneolignans†
Mukesh Gangar,
Avinash Ittuveetil,
Sandeep Goyal,
Anang Pal,
Harikrishnan M. and
Vipin A. Nair*
Department of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research, Sector 67, Mohali, Punjab 160062, India. E-mail: vn74nr@yahoo.com; Fax: +91-172-2214692
First published on 14th October 2016
Abstract
The anti selective glycolate aldol reactions of (S)-4-isopropyl-1-[(R)-1-phenylethyl]imidazolidin-2-one auxiliary have been standardized with high yields and excellent diastereoselectivities on various substituted aryl, allyl and alkyl aldehydes. The optimized reaction conditions were employed for the stereoselective synthesis of oxyneolignans.
Introduction
The glycolate aldol reaction is a convenient method to access 1,2-diols, and serves as an attractive alternative to the dihydroxylation of double bonds.1 Through energetically differing transition states, stereoselective aldol reactions offer the advantage of yielding one of the isomers preferentially. Chiral auxiliary mediated glycolate aldol reactions provide a robust method for the stereoselective synthesis of 1,2-diols,2 (Fig. 1) while only a few chiral catalysts and ligands are yet known to promote the reaction.3 Glycolate esters of oxazolidinone and oxazolidinethione based chiral auxiliaries have been employed to demonstrate syn 1,2-diol formation by the intermediacy of boron and titanium enolates, respectively.2c,2k On the contrary, the anti selective 1,2-diol formation is innately more challenging, and is assumed to be due to the difficulty in attaining an E-enolate geometry in the transition state with many of the chiral auxiliaries. The available information from literature indicate that only a few chiral auxiliaries are known to afford the anti 1,2-diols, with inadequate scope for a broad substrate generalization.2d,2g,2h,2i Among them, the tin enolates of oxazolidinone and thiazolidinone had revealed a biasness for the formation of anti glycolate aldol adducts, though the yield/selectivity was insufficient.2f The stereoselective glycolate aldol reactions of boron enolate of a chiral oxapyrone exhibited anti diastereoselectivity.2g The reaction employed stoichiometric excess of the reagents for enolization, affording only moderate yields and stereoselectivities with aromatic and α,β-unsaturated aldehydes. With a substrate limitation of the presence of an alkoxy group on aldehydes, and a condition of precomplexation with TiCl4, high anti diastereoselectivity was illustrated in glycolate aldol reactions involving titanium enolates of N-glycolylselones.2i Another improvement in this direction was demonstrated with the oxazolidinethione glycolate. Despite the reasonably good anti diastereoselectivity obtained in the reactions with aliphatic aldehydes, titanium enolate of the N-glycolyloxazolidinethione also necessitated the precomplexation of aldehydes with excess of TiCl4.2h The boron enolate generated from the glycolate ester of the benzyl protected thiol variant of a norephedrine-derived chiral auxiliary illustrated anti diastereoselectivity.2d However the facial selectivity was unsatisfactory, and a switch over to tert-butyldiphenylsilyl protecting group was essential. Imposed by the condition employed for the cleavage of the auxiliary from the aldol adduct, in some cases the limitations also included the difficulty in recovering the chiral auxiliary for further use. The predicaments demand an alternative for the synthesis of anti 1,2-diols. Recently we observed a reversal in selectivity from syn to anti aldol adducts in the acetate aldol reactions of an L-valine derived chiral auxiliary, (S)-4-isopropyl-1-(R)-1-phenylethyl]imidazolidin-2-one by changing the enolate metal counter ion from titanium to lithium.4 Perceptions in this direction prompted us to explore the glycolate aldol reactions of the auxiliary by generating the lithium enolate. We reasoned that the attempt would provide an easy access to the stereochemically defying anti 1,2-diol synthesis. Elaboration of the use of the imidazolidin-2-one chiral auxiliary to variations of the aldol reaction theme would also exemplify its versatility, the stereoselectivity arising from the dual stereochemical controls consisting of the isopropyl group residing on the heterocycle that induces the facial biasness, and the exocyclic α-methyl benzyl substituent that augments the stereoselection by a synchronous electronic effect.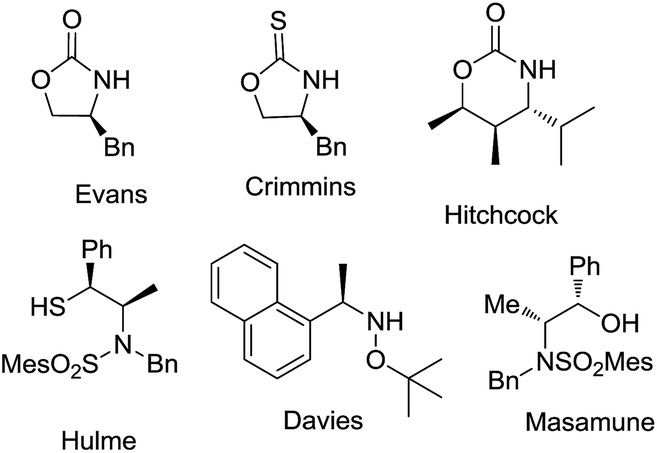 | ||
| Fig. 1 A few auxiliaries used for glycolate aldol reaction. | ||
The N-acylation reaction was carried out by the deprotonation of the chiral auxiliary (S)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one 1 with NaH in freshly dried THF followed by the treatment with benzyloxyacetyl chloride to afford the acylated chiral auxiliary 2 in 82% yield. Evaluation of the aldol reaction was carried out by generating the lithium enolate of benzyl protected N-glycolyl-(S)-4-isopropyl-1-(R)-1-phenylethyl]imidazolidin-2-one (Scheme 1). Among the different lithium bases employed for enolate generation LiHMDS afforded the best result. A preliminary reaction with benzaldehyde afforded high diastereoselectivity of 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :02 in favor of the anti glycolate aldol adduct as determined from the 1H NMR spectrum of the crude product. The scope of the reaction was examined by extending the reaction condition to different aldehydes and excellent anti diastereoselectivity was obtained in all the cases, including aromatic, heteroaromatic and α,β-unsaturated aldehydes (Table 1).
:02 in favor of the anti glycolate aldol adduct as determined from the 1H NMR spectrum of the crude product. The scope of the reaction was examined by extending the reaction condition to different aldehydes and excellent anti diastereoselectivity was obtained in all the cases, including aromatic, heteroaromatic and α,β-unsaturated aldehydes (Table 1).
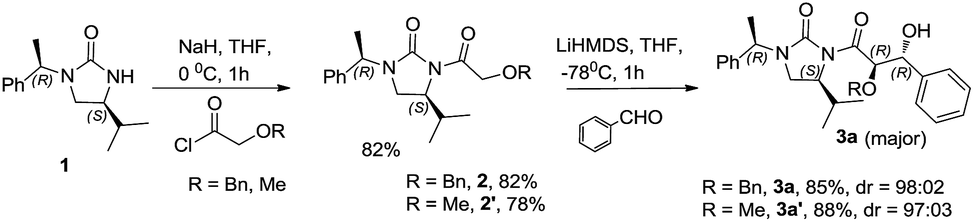 | ||
| Scheme 1 Glycolate aldol reaction of (S)-4-isopropyl-1-(R)-1-phenylethyl]imidazolidin-2-one. | ||
| Entry | Substrate | Product | Yield (%) | dr (anti 1:∑anti 2, syn) |
|---|---|---|---|---|
| a Diastereomeric ratio was determined from the 1H NMR spectrum of the reaction mixture. | ||||
| 1 | Benzaldehyde | 3a | 85 | 98:2 |
| 2 | 4-Fluorobenzaldehyde | 3b | 86 | 98:2 |
| 3 | 4-Chlorobenzaldehyde | 3c | 88 | 98:2 |
| 4 | 4-Bromobenzaldehyde | 3d | 79 | 98:2 |
| 5 | 4-Methylbenzaldehyde | 3e | 78 | 96:4 |
| 6 | 2-Methylbenzaldehyde | 3f | 75 | 93:7 |
| 7 | Furan-2-carboxaldehyde | 3g | 71 | 98:2 |
| 8 | 2-Methylbut-2-enal | 3h | 80 | 95:5 |
| 9 | Isovaleraldehyde | 3i | 70 | 95:5 |
| 10 | Cinnamaldehyde | 3j | 76 | 96:4 |
| 11 | Propionaldehyde | 3k | 82 | 97:3 |
| 12 | 4-OTBS vanillin | 3l | 85 | 96:4 |
| 13 | 4-OTBS syringaldehyde | 3m | 82 | 95:5 |
| 14 | Isobutyraldehyde | 3n | 78 | 96:4 |
To determine the absolute configuration aldol adduct 3l was reductively cleaved and subsequently deprotected to give 4 in 75% isolated yield over 3 steps (Scheme 2). The diastereoselectivity observed can be explained by the transition states depicted in Scheme 3. Reaction of the acylated chiral auxiliary with LiHMDS is expected to generate the (Z) enolate preferentially. Attack of the enolate from its Re face to the Re face of the aldehyde is energetically favored over the Si face of the aldehyde, through transition state (TS-A) to afford the glycolate anti aldol isomer as the major product.
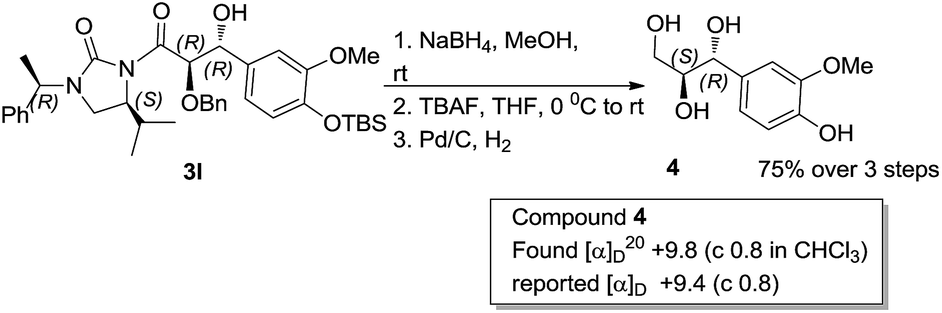 | ||
| Scheme 2 Determination of absolute configuration. | ||
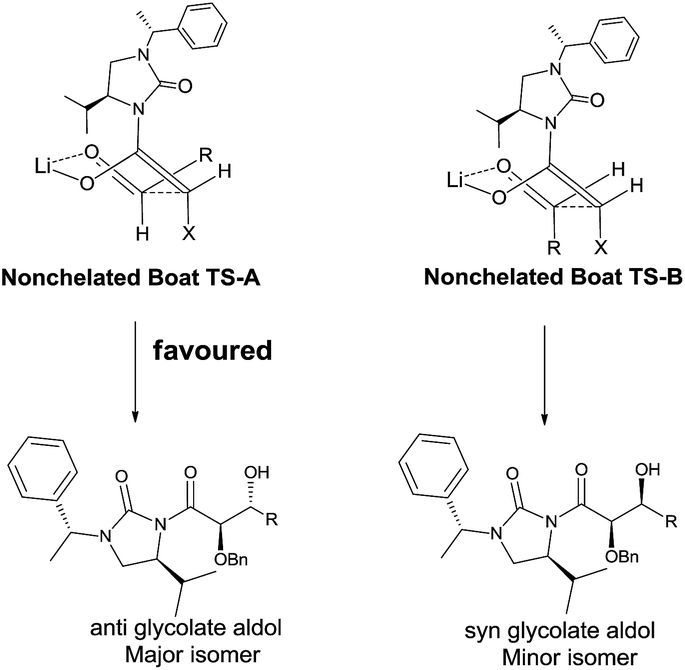 | ||
| Scheme 3 Possible transition state for the lithium mediated glycolate aldol reaction. | ||
Cleavage of the auxiliary from the aldol adduct under different conditions afforded the functionally modified 1,2-diols. For instance, sodium borohydride mediated reduction in methanol gave the synthetically useful propane-1,2,3-triol unit 7 wherein one of the secondary hydroxyl groups is selectively protected by a benzyl group. Such a selective protection on 1,2,3-triols is normally difficult to achieve. Similarly, reactions of the aldol adducts with amines and alcohols yielded the amide and ester derivatives of 1,2-diols, respectively. The mild procedure of subjecting an aldol intermediate to hydrogen peroxide in presence of a base revealed the corresponding carboxylic acid (Scheme 4). In all these cases the chiral auxiliary was successfully recovered in good yields (85–88%) by column chromatographic methods, for recycling. The anti 1,2-diol selectivity and absolute configuration were ascertained by comparing the optical rotation of compound 4 with that of the literature and also by single crystal X-ray diffraction analysis of compound 3a (Fig. 2).
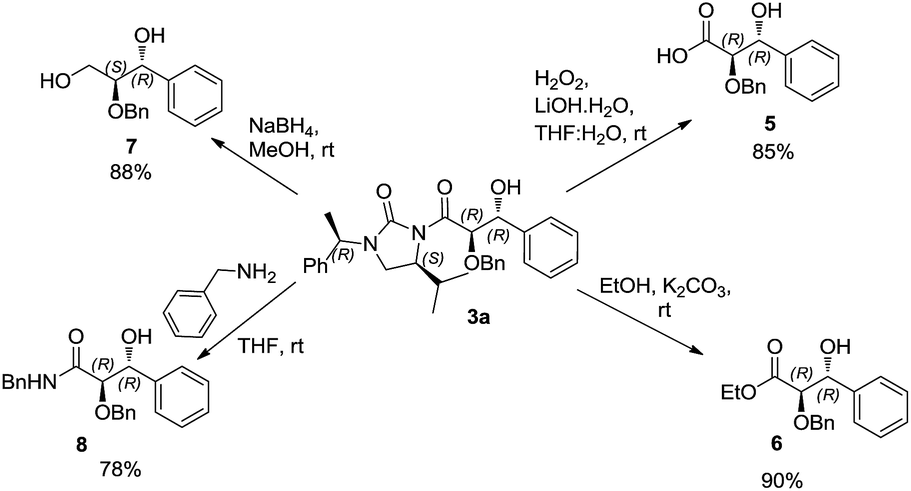 | ||
| Scheme 4 Cleavage of the chiral auxiliary. | ||
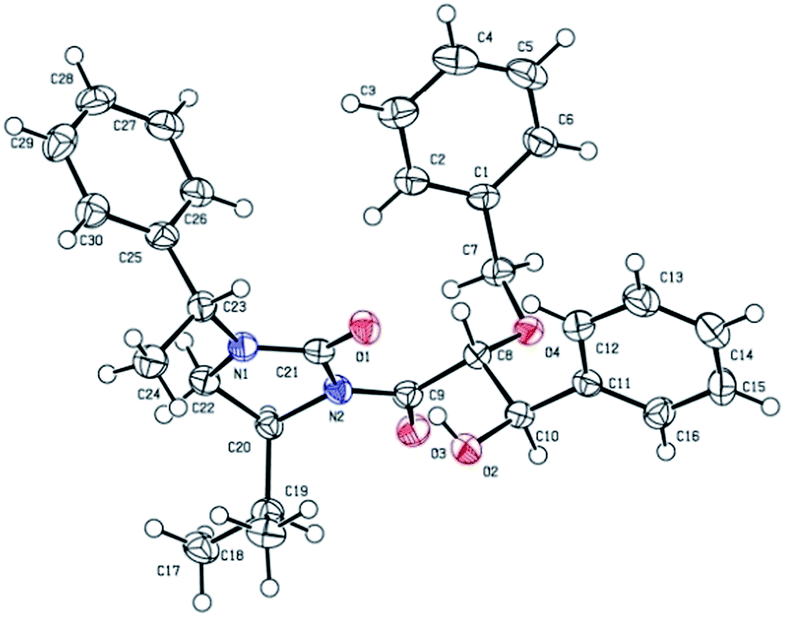 | ||
| Fig. 2 Ortep diagram of compound 3a. | ||
Encouraged by the high anti diastereoselectivity obtained in the glycolate aldol reactions we decided to extend the strategy towards the synthesis of 8-4′-oxyneolignans (Scheme 5). Lignans, the products of shikimic acid pathway, encompass compounds containing two phenyl propanoid (C6–C3) units joined together at C8 and C8′. Neolignans on the other hand, are composed of two C6–C3 units which are linked via carbon atoms other than C8 while the subclass of oxyneolignans have their C6–C3 units not directly joined but connected via an ether linkage. The locants of the two positions linked by the ether oxygen are cited in front of the name with the second number primed.4 The 8-4′-oxyneolignans have become the subject of extensive research lately due to their vast pharmacological profile, which includes antimalarial, antiparasitic, antifungal, antioxidant, antileishmanial, anti-schistosomial and anticancer properties (Fig. 3).5,6
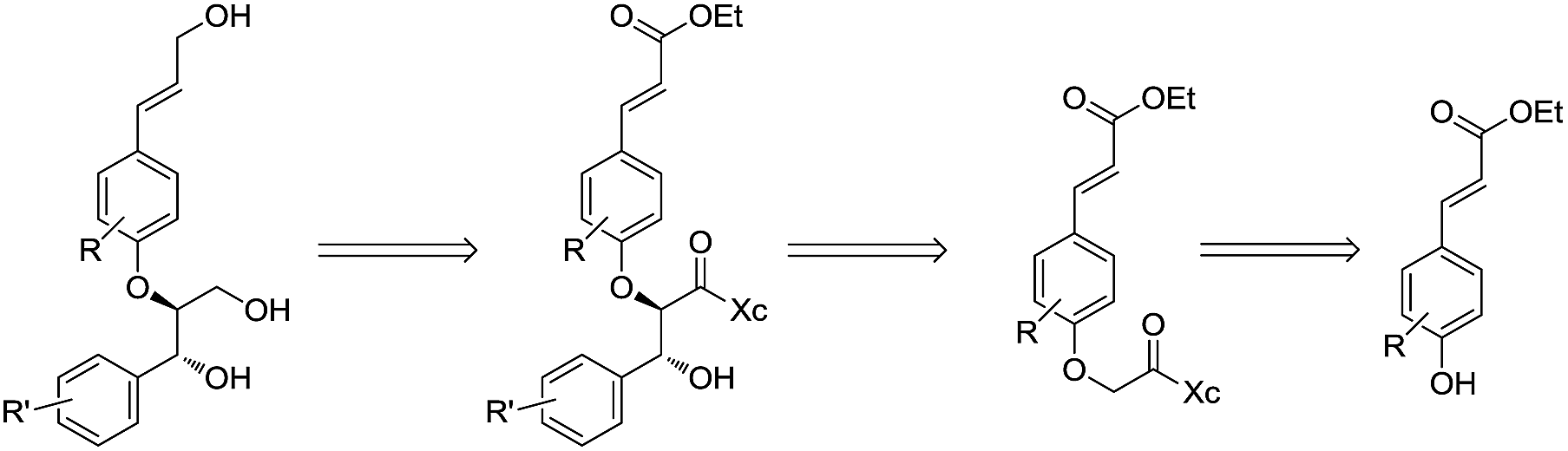 | ||
| Scheme 5 Retrosynthetic scheme for 8-4′-oxyneolignans. | ||
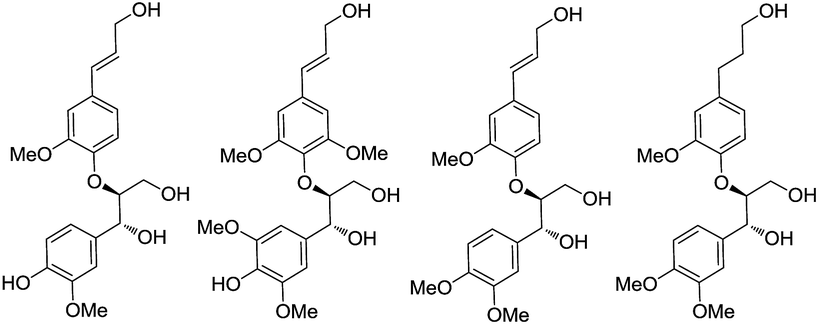 | ||
| Fig. 3 Structurally diverse oxyneolignans. | ||
Wide range of substituents have been observed in these molecules which contribute to the diverse chemical features and biological activities. 8-4′-Oxyneolignans having 3-hydroxy-(E)-prop-1-enyl side chain in the erythro and threo forms and 3-hydroxy propyl side chains are found in several natural sources. Despite the difficulties in the isolation of this pharmacologically active ingredient from the natural sources, no stereoselective synthetic procedures have been reported till date. Taking further strides, we explored a suitable retrosynthetic pathway for the asymmetric synthesis of the template. The synthesis of the guiacol ether commenced with a silyl protection of 4-hydroxy-3,5-dimethoxybenzaldehyde (syringaldehyde) using tert-butyldimethylsilyl chloride and imidazole in DCM at 0 °C (Scheme 6). The protected benzaldehyde 9 was subjected to Horner–Wadsworth–Emmons reaction employing triethylphosphonoacetate and DBU at room temperature under neat conditions to obtain the corresponding conjugated ester. The silyl protection was then removed using DBU in acetonitrile-water mixture at room temperature to give ethyl sinapate 10. Similarly the monomethoxy analogue ethyl ferulate 11 was synthesized from commercially available ferulic acid by Fischer esterification.
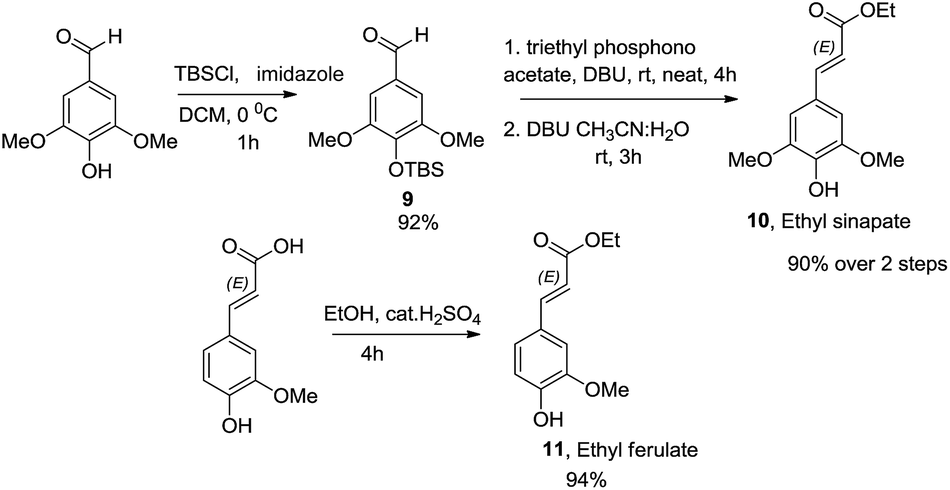 | ||
| Scheme 6 Synthesis of ethyl sinapate and ethyl ferulate. | ||
The esters 10 & 11 were converted to the corresponding alcohols using a combination of lithium aluminium hydride and benzyl chloride in THF at 0 °C to obtain coniferyl alcohol 13a and sinapyl alcohol 13b, respectively. The bromoacetyl derivative of (S)-4-isopropyl-1-[(R)-1-phenylethyl]imidazol-idin-2-one was prepared by deprotonation of the chiral auxiliary 1 with NaH in dry THF followed by treatment with bromoacetyl bromide. Reaction of 13a & 13b with the bromoacetylated chiral auxiliary 12 and potassium carbonate in acetone along with the catalytic amount of KI at room temperature afforded the corresponding aryloxy acetyl derivatives. Subsequently the allylic alcohol was protected using tert-butyldimethylsilyl chloride and thereafter subjected to enolization using LiHMDS in THF at −78 °C. The reaction of the corresponding lithium enolate with the different aldehydes afforded the anti aldol derivatives 15(a–c). The chiral auxiliary was removed by a reductive cleavage employing NaBH4 in THF–H2O medium at room temperature. Reaction with tetrabutylammonium fluoride in THF at room temperature afforded the corresponding oxyneolignans 16(a–c) (Scheme 7). The allylic side chain of oxyneolignan 16a was reduced by catalytic hydrogenation using Pd/C in ethyl acetate at room temperature to give the corresponding oxyneolignan 17. The anti glycolate aldol stereochemistry was confirmed by comparing the coupling constant (reported J7,8 = 3.5 Hz, observed J7,8 = 3.5 Hz). The absolute configuration was established by correlation of the optical rotation [reported [α]18D 7.7 (c 1.9, MeOH), observed [α]20D 9.2 (c 1.9, MeOH)].
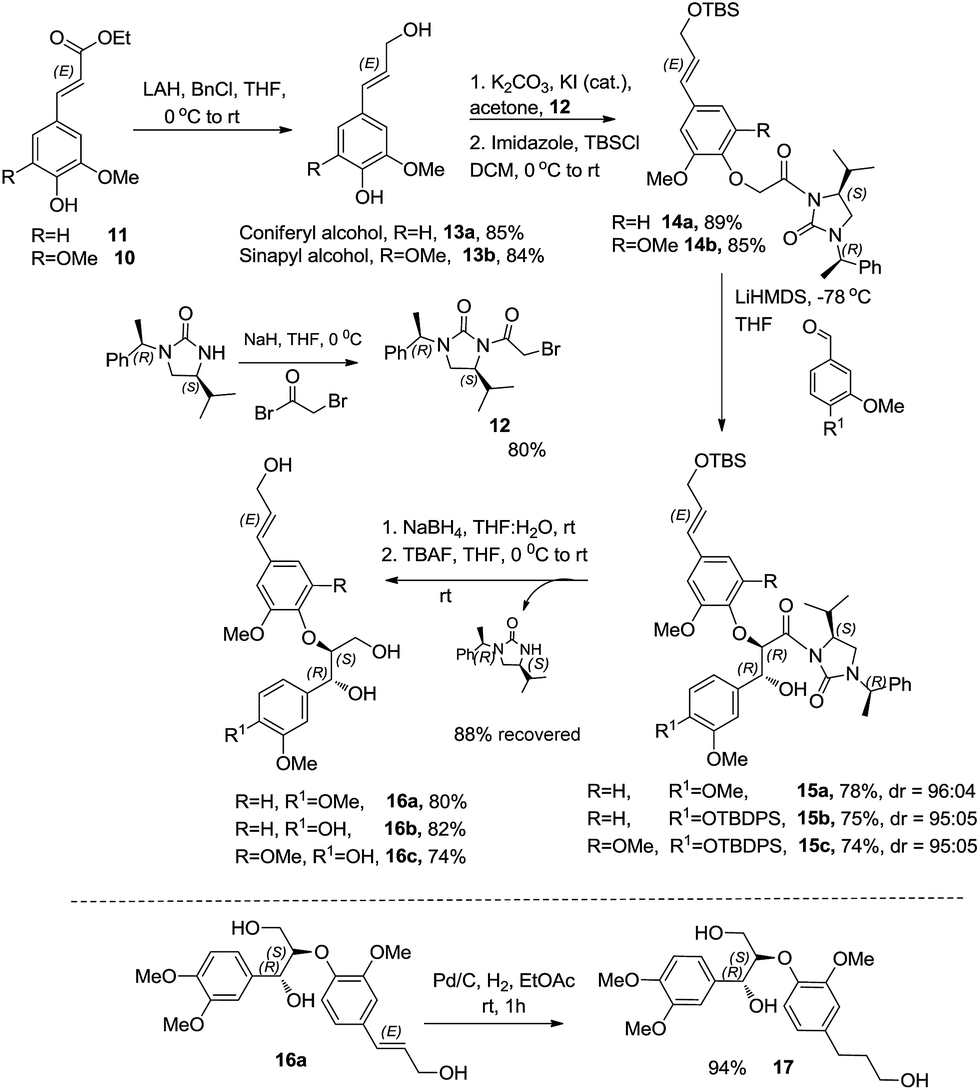 | ||
| Scheme 7 Total synthesis of oxyneolignans. | ||
Conclusions
The anti glycolate aldol reaction of benzyloxy acetyl derivatives of (S)-4-isopropyl-1-(R)-1-phenylethyl]imidaz-olidin-2-one has been successfully standardised with high yields and diastereoselectivity. Further its application in the total synthesis of naturally occurring 8-4′-oxyneolignans was demonstrated.Experimental
All the reagents required for this study were purchased from commercial sources and used as such without further purification. Solvents were distilled and dried before use. 1H and 13C NMR spectra were recorded at 400 and 100 MHz, respectively, on a Bruker Avance DPX 400 (400 MHz) spectrometer in CDCl3/CD3OD using TMS as an internal standard. The chemical shifts (δ) for 1H and 13C spectra are given in ppm relative to residual signals of the solvent. Coupling constants are given in Hz. The following abbreviations are used to indicate multiplicity: s, singlet; d, doublet; t, triplet; td, triple doublet; dt, double triplet; q, quartet; m, multiplet; brs, broad signal. HRMS were recorded on a Bruker Maxix TOF mass spectrometer. Melting points are uncorrected. IR spectra were recorded on a Perkin Elmer FTIR spectrophotometer.Crystallographic data for the compound 3a has been deposited with the Cambridge Crystallographic Data Centre, CCDC no. 1452671.
Synthesis of (S)-3-(2-(benzyloxy)acetyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (2a)
To a stirring solution of (S)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (2.0 g, 8.6 mmol, 1.0 equiv.) in anhydrous THF, sodium hydride (60% suspension in paraffin oil, 0.41 g, 10.3 mmol, 1.2 equiv.) was slowly added at 0 °C under inert atmosphere. After 1 h, benzyloxyacetyl chloride (1.43 mL, 9.0 mmol, 1.05 equiv.) was added dropwise. Reaction mixture was stirred for another hour. Completion of the reaction was monitored by TLC. The reaction mixture was then quenched by dropwise addition of aq. NH4Cl at 0 °C, washed with water and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate. It was evaporated under vacuum to give a solid compound which was purified by column chromatography on silica gel (60–120 mesh) using ethyl acetate–hexane (01:09) mixture as eluent.
Yield = 82%, white solid, mp = 69 °C, [α]20D = +89.1 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.81 (d, J = 6.8 Hz, 3H), 0.88 (d, J = 6.8 Hz, 3H), 1.55 (d, J = 7.1 Hz, 1H), 2.42–2.50 (m, 1H), 2.96 (t, J = 9.6 Hz, 1H), 3.10 (dd, J = 3.0, 9.6 Hz, 1H), 4.18–4.22 (m, 1H), 4.66 (q, J = 11.7 Hz, 1H), 4.76 (d, J = 1.9 Hz, 1H), 5.28 (q, J = 7.1 Hz, 1H), 7.28–7.36 (m, 8H), 7.40–7.42 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 14.39, 16.12, 18.01, 28.48, 37.81, 50.32, 54.87, 70.03, 73.41, 127.15, 127.78, 128.00, 128.07, 128.41, 128.77, 137.70, 138.83, 154.40, 170.47; ESI-HRMS (m/z): [M + Na]+ calculated for C23H28N2NaO3, 403.1998, found, 403.1998.
(S)-4-Isopropyl-3-(2-methoxyacetyl)-1-((R)-1-phenylethyl)imidazolidin-2-one (2a′)
White solid; yield: 78%; mp: 88 °C; [α]20D = +110.1 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.82 (d, J = 6.9 Hz, 3H), 0.89 (d, J = 7.0 Hz, 3H), 1.57 (d, J = 7.2 Hz, 3H), 2.41–2.49 (m, 1H), 2.98 (t, J = 9.5 Hz, 1H), 3.12 (dd, J = 3.0, 9.6 Hz, 1H), 3.49 (s, 3H), 4.21 (dt, J = 3.3, 9.4 Hz, 1H), 4.65 (d, J = 17.5 Hz, 1H), 4.71 (d, J = 17.5 Hz, 1H), 5.30 (q, J = 7.1 Hz, 1H), 7.29–7.39 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 14.44, 16.13, 17.99, 28.52, 37.90, 50.36, 54.82, 59.36, 72.43, 127.14, 128.78, 128.01, 138.87, 154.44, 170.32; IR (cm−1): ν = 3444, 2963, 1719, 1495, 1428, 1304, 1262, 1202, 1130, 933, 891, 764, 702, 647, 547; HRMS (ESI-TOF) calcd for C17H24N2O3Na [M + Na]+: 327.1685; found: 327.1671.General procedure for the aldol reaction
To the solution of (S)-3-[2-(benzyloxy)acetyl]-4-isopropyl-1-[(R)-1-phenylethyl]imidazolidin-2-one (3) (0.6 g, 2.2 mmol, 1.0 equiv.) in freshly dried THF (5 mL) under N2 environment and cooled to −78 °C was added LiHMDS solution (2.4 mL, 2.4 mmol, 1.1 equiv.; 1 M in THF) at −78 °C, and stirred for 1 h. Benzaldehyde (0.25 mL, 2.4 mmol, 1.1 equiv.) was added to the reaction mixture and further stirred for 30 minutes. The reaction was quenched by saturated aq. NH4Cl solution, and extracted with EtOAc which was further washed with brine and dried over anhydrous sodium sulfate. The crude reaction mixture was purified by column chromatography using silica gel (60–120 mesh) by eluting with petroleum ether/EtOAc (60:40) to afford the desired product (0.77 g, 85%).
(S)-3-((2R,3R)-2-(Benzyloxy)-3-hydroxy-3-phenylpropanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3a)
Yield = 85%; crystalline solid, mp = 85–86 °C. [α]20D = +99.7 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.75 (d, J = 6.9 Hz, 3H), 0.87 (d, J = 7.0 Hz, 3H), 1.59 (d, J = 7.2 Hz, 3H), 2.33–2.41 (m, 1H), 2.88 (t, J = 9.4 Hz, 1H), 3.08 (dd, J = 2.3, 9.7 Hz, 1H), 3.98–4.02 (m, 1H), 4.08 (d, J = 9.8 Hz, 1H), 4.34 (d, J = 11.8 Hz, 1H), 4.42 (d, J = 11.8 Hz, 1H), 4.85 (dd, J = 8.3, 9.7 Hz, 1H), 5.35 (q, J = 7.2 Hz, 1H), 5.52 (d, J = 8.2 Hz, 1H), 7.06–7.17 (m, 5H), 7.30–7.47 (m, 8H), 7.52–7.55 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 14.47, 16.06, 17.99, 28.78, 37.66, 50.60, 55.79, 72.98, 75.21, 79.39, 126.97, 127.26, 127.64, 127.73, 128.06, 128.18, 128.26, 128.47, 128.81, 137.29, 138.74, 141.95, 155.12, 171.75; ESI-HRMS (m/z): [M + Na]+ calculated for C30H34N2NaO4, 509.2416, found, 509.2416.(S)-3-((2R,3R)-3-Hydroxy-2-methoxy-3-phenylpropanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3a′)
Yield = 88%; gummy. [α]20D = +74.3 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.77 (d, J = 6.9 Hz, 3H), 0.90 (d, J = 7.0 Hz, 3H), 1.61 (d, J = 7.2 Hz, 3H), 2.36–2.44 (m, 1H), 3.01 (t, J = 9.4 Hz, 1H), 3.14 (dd, J = 2.4, 9.8 Hz, 1H), 3.25 (s, 3H), 4.04 (d, J = 8.4 Hz, 1H), 4.28–4.32 (m, 1H), 4.83 (t, J = 8.4 Hz, 1H), 5.40 (q, J = 7.1 Hz, 1H), 5.43 (d, J = 7.7 Hz, 1H), 7.30–7.42 (m, 8H), 7.53 (d, J = 7.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.37, 16.10, 18.03, 28.68, 37.64, 55.71, 58.31, 74.99, 77.25, 81.78, 126.86, 127.20, 127.80, 128.15, 128.32, 128.85, 138.60, 141.71, 155.17, 171.48; ESI-HRMS (m/z): [M + Na]+ calculated for C24H30N2NaO4, 433.2103, found, 433.2101.(S)-3-((2R,3R)-2-(Benzyloxy)-3-(4-fluorophenyl)-3-hydroxy propanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3b)
Yield = 86%; gummy. [α]20D = +94.3 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.73 (d, J = 6.9 Hz, 3H), 0.85 (d, J = 7.0 Hz, 3H), 1.56 (d, J = 7.2 Hz, 3H), 2.29–2.37 (m, 1H), 2.86 (t, J = 9.4 Hz, 1H), 3.06 (dd, J = 11.9 Hz, 1H), 3.95–3.99 (m, 1H), 4.15 (d, J = 9.1 Hz, 1H), 4.32 (d, J = 11.8 Hz, 1H), 4.38 (d, J = 11.8 Hz, 1H), 4.81 (t, J = 8.6 Hz, 1H), 5.31 (q, J = 7.1 Hz, 1H), 5.43 (d, J = 8.1 Hz, 1H), 7.01–7.10 (m, 6H), 7.11–7.16 (m, 1H), 7.31–7.48 (m, 7H); 13C NMR (100 MHz, CDCl3) δ 14.47, 16.08, 17.97, 28.82, 37.71, 50.67, 55.83, 72.98, 74.55, 79.36, 114.90, 115.11, 127.25, 127.74, 128.10, 128.23, 128.50, 128.64, 128.72, 128.84, 137.12, 137.87, 137.90, 138.69, 155.14, 161.18, 163.62, 171.56; ESI-HRMS (m/z): [M + Na]+ calculated for C30H33FN2NaO4, 527.2322, found, 527.2326.(S)-3-((2R,3R)-2-(Benzyloxy)-3-(4-chlorophenyl)-3-hydroxy propanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3c)
Yield 88%; gummy. [α]20D = +88.9 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.75 (d, J = 6.9 Hz, 3H), 0.87 (d, J = 7.0 Hz, 3H), 1.59 (d, J = 7.2 Hz, 3H), 2.31–2.39 (m, 1H), 2.88 (t, J = 9.4 Hz, 1H), 3.09 (dd, J = 2.2, 9.7 Hz, 1H), 3.97–4.01 (m, 1H), 4.23 (d, J = 9.7 Hz, 1H), 4.35 (d, J = 11.8 Hz, 1H), 4.42 (d, J = 11.8 Hz, 1H), 4.82 (t, J = 9.0 Hz, 1H), 5.34 (q, J = 7.1 Hz, 1H), 5.44 (d, J = 8.2 Hz, 1H), 7.05–7.19 (m, 5H), 7.33–7.36 (m, 4H), 7.37–7.47 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 14.44, 16.08, 17.97, 28.83, 37.71, 50.69, 55.84, 73.02, 74.54, 79.20, 127.25, 127.77, 128.11, 128.25, 128.34, 128.40, 128.55, 128.85, 133.43, 137.02, 138.65, 140.57, 155.16, 171.47; ESI-HRMS (m/z): [M + Na]+ calculated for C30H33ClN2NaO4, 543.2027, found, 543.2029.(S)-3-((2R,3R)-2-(Benzyloxy)-3-(4-bromophenyl)-3-hydroxy propanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3d)
Yield = 79%; gummy. [α]20D = +73.2 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.72 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 7.0 Hz, 3H), 1.57 (d, J = 7.2 Hz, 3H), 2.29–2.37 (m, 1H), 2.86 (t, J = 9.4 Hz, 1H), 3.06 (dd, J = 2.1, 9.7 Hz, 1H), 3.94–3.98 (m, 1H), 4.19 (d, J = 8.9 Hz, 1H), 4.32 (d, J = 11.8 Hz, 1H), 4.39 (d, J = 11.8 Hz, 1H), 4.78 (t, J = 7.6 Hz, 1H), 5.31 (q, J = 7.1 Hz, 1H), 5.40 (d, J = 8.2 Hz, 1H), 7.07 (d, J = 4.3 Hz, 4H), 7.12–7.17 (m, 1H), 7.29–7.49 (m, 9H); 13C NMR (100 MHz, CDCl3) δ 14.44, 16.07, 17.96, 28.84, 37.73, 50.69, 55.84, 73.01, 74.59, 79.15, 121.64, 127.24, 127.77, 128.12, 128.24, 128.54, 128.73, 128.84, 131.28, 137.01, 138.65, 141.07, 155.17, 171.41; ESI-HRMS (m/z): [M + Na]+ calculated for C30H33BrN2NaO4, 587.1521, found, 587.1531.(S)-3-((2R,3R)-2-(Benzyloxy)-3-hydroxy-3-(p-tolyl)propanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3e)
Yield = 78%; gummy. [α]20D = +86.1 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.76 (d, J = 6.9 Hz, 3H), 0.88 (d, J = 7.0 Hz, 3H), 1.59 (d, J = 7.2 Hz, 3H), 2.34–2.41 (m, 4H; Me, s, 3H & m, 1H merged), 2.87 (t, J = 9.4 Hz, 1H), 3.08 (dd, J = 2.2, 9.6 Hz, 1H), 3.97–4.02 (m, 2H), 4.35 (d, J = 11.8 Hz, 1H), 4.45 (d, J = 11.8 Hz, 1H), 4.83 (t, J = 9.1 Hz, 1H), 5.36 (q, J = 7.1 Hz, 1H), 5.54 (d, J = 8.2 Hz, 1H), 7.07–7.18 (m, 5H), 7.21 (d, J = 7.9 Hz, 2H), 7.33–7.40 (m, 3H), 7.42–7.46 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 14.47, 16.09, 18.01, 21.23, 28.80, 37.62, 50.60, 55.76, 73.01, 75.05, 79.46, 126.90, 127.28, 127.62, 128.04, 128.19, 128.51, 128.82, 128.95, 137.29, 137.39, 138.79, 139.04, 155.09, 171.88; ESI-HRMS (m/z): [M + Na]+ calculated for C31H36N2NaO4, 523.2573, found, 523.2577.(S)-3-((2R,3R)-2-(Benzyloxy)-3-hydroxy-3-(o-tolyl)propanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3f)
Yield = 75%; gummy. [α]20D = +95.1 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.74 (d, J = 6.9 Hz, 3H), 0.89 (d, J = 7.0 Hz, 3H), 1.58 (d, J = 7.1 Hz, 3H), 2.38–2.47 (m, 4H; Me, 3H & m, 1H, merged), 2.88 (t, J = 9.4 Hz, 1H), 3.08 (dd, J = 2.2, 9.7 Hz, 1H), 4.02–4.06 (m, 2H) 4.39 (s, 2H), 5.20 (t, J = 8.8 Hz, 1H), 5.36 (q, J = 7.1 Hz, 1H), 5.67 (d, J = 8.2 Hz, 1H), 7.10 (d, J = 4.4 Hz, 4H), 7.14–7.29 (m, 4H), 7.33–7.40 (m, 3H), 7.42–7.46 (m, 2H), 7.62 (d, J = 7.4 Hz, 1H); 13C 14.50, 16.07, 18.05, 19.56, 28.81, 37.54, 50.58, 55.76, 71.23, 72.89, 79.13, 125.85, 126.24, 127.28, 127.46, 127.61, 128.06, 128.18, 128.33, 128.83, 130.16, 136.64, 137.46, 138.79, 140.31, 154.99, 172.04; ESI-HRMS (m/z): [M + Na]+ calculated for C31H36N2NaO4, 523.2573, found, 523.2561.(S)-3-((2R,3S)-2-(Benzyloxy)-3-(furan-2-yl)-3-hydroxypropanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3g)
Yield = 71%; gummy. [α]20D = +117.7 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.80 (d, J = 6.9 Hz, 3H), 0.88 (d, J = 7.0 Hz, 3H), 1.59 (d, J = 7.2 Hz, 3H), 2.36–2.44 (m, 1H), 2.90 (t, J = 9.4 Hz, 1H), 3.09 (dd, J = 2.4, 9.7 Hz, 1H), 3.90 (d, J = 10.3 Hz, 1H), 4.01–4.05 (m, 1H), 4.48 (s, 2H), 4.91 (t, J = 9.2 Hz, 1H), 5.38 (q, J = 7.1 Hz, 1H), 5.69 (d, J = 8.2 Hz, 1H), 6.38 (dd, J = 1.8, 3.2 Hz, 1H), 6.42 (d, J = 3.1 Hz, 1H), 7.12–7.23 (m, 5H), 7.35–7.39 (m, 3H), 7.42–7.45 (m, 3H); 13C NMR 14.43, 16.04, 17.96, 28.70, 37.68, 50.58, 53.76, 68.71, 73.12, 77.68, 108.12, 110.34, 127.29, 127.74, 128.15, 128.48, 128.81, 137.73, 142.18, 154.13, 154.96, 171.13; ESI-HRMS (m/z): [M + Na]+ calculated for C28H32N2NaO5, 499.2209, found, 499.2209.(S)-3-((2R,3R,E)-2-(Benzyloxy)-3-hydroxy-4-methylhex-4-enoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3h)
Yield = 80%; gummy. [α]20D = +107.7 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.81 (d, J = 6.9 Hz, 3H), 0.88 (d, J = 7.0 Hz, 3H), 1.59 (d, J = 6.7 Hz, 3H), 1.67 (d, J = 6.7 Hz, 3H), 1.72 (s, 3H), 2.34–2.42 (m, 1H), 2.88 (t, J = 9.4 Hz, 1H), 3.08 (dd, J = 2.3, 9.6 Hz, 1H), 3.58 (d, J = 9.0 Hz, 1H), 3.99–4.03 (m, 1H), 4.23 (t, J = 7.6 Hz, 1H), 4.53 (s, 2H), 5.35 (q, J = 7.2 Hz, 1H), 5.43 (d, J = 8.0 Hz, 1H), 5.63 (q, J = 6.7 Hz, 1H), 7.13–7.20 (m, 3H), 7.26–7.28 (m, 2H), 7.35–7.39 (m, 3H), 7.42–7.46 (m, 2H); 13C NMR 11.41, 13.32, 14.47, 16.07, 17.97, 28.76, 37.56, 50.55, 55.66, 72.88, 76.80, 78.45, 123.48, 127.28, 127.71, 128.12, 128.16, 128.50, 128.80, 135.01, 137.55, 138.81, 155.00, 172.12; HRMS (ESI-TOF) calculated for C28H32N2O4Na [M + Na]+: 487.2573; found: 487.2573.(S)-3-((2R,3R)-2-(Benzyloxy)-3-hydroxy-5-methylhexanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3i)
Yield = 70%; gummy. [α]20D = +67.9 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ (400 MHz, CDCl3) 0.73 (d, J = 6.9 Hz, 3H), 0.79 (d, J = 7.0 Hz, 3H), 0.84 (d, J = 6.6 Hz, 3H), 0.88 (d, J = 6.7 Hz, 3H), 1.45 (t, J = 6.8 Hz, 2H), 1.49 (d, J = 7.2 Hz, 3 h), 1.82–1.89 (m, 1H), 2.27–2.34 (m, 1H), 2.62 (d, J = 10.1 Hz, 1H), 2.82 (t, J = 9.4 Hz, 1H), 3.01 (dd, J = 2.3, 9.6 Hz, 1H), 3.82–3.89 (m, 1H), 3.93–3.97 (m, 1H), 4.46 (s, 2H), 5.11 (d, J = 6.6 Hz, 1H), 5.23 (d, J = 7.1 Hz, 1H), 7.07–7.12 (m, 2H), 7.18–7.36 (m, 8H); 13C NMR (100 MHz, CDCl3) 14.53, 16.12, 17.98, 21.53, 23.98, 24.30, 28.74, 37.62, 43.47, 50.62, 55.51, 71.31, 72.98, 80.76, 127.25, 127.76, 128.07, 128.18, 128.60, 128.81, 137.51, 138.85; HRMS (ESI-TOF) calculated for C32H36N2O4Na [M + Na]+: 535.2573; found: 535.2576.(S)-3-((2R,3R,E)-2-(Benzyloxy)-3-hydroxy-5-phenylpent-4-enoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3j)
Yield = 76%; gummy. [α]20D = +136.3 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.75 (d, J = 6.9 Hz, 3H), 0.86 (d, J = 7.1 Hz, 3H), 1.58 (d, J = 7.2 Hz, 3H), 2.31–2.40 (m, 1H), 2.91 (t, J = 9.4 Hz, 1H), 3.09 (dd, J = 2.4, 9.6 Hz, 1H), 3.48 (d, J = 9.4 Hz, 1H), 4.02–4.06 (m, 1H), 4.60 (s, 2H), 5.34 (q, J = 7.7 Hz, 1H), 5.41 (d, J = 6.8 Hz, 1H), 6.48 (dd, J = 5.6, 16.0 Hz, 1H), 6.76 (d, J = 15.4 Hz, 1H), 7.08–7.28 (m, 4H), 7.31–7.46 (m, 11H); 13C NMR (100 MHz, CDCl3) δ 14.45, 16.11, 17.98, 28.76, 37.67, 50.67, 55.64, 73.08, 73.35, 79.39, 126.68, 127.26, 127.52, 127.86, 128.19, 128.24, 128.45, 128.66, 128.83, 128.91, 131.27, 136.89, 137.35, 138.80, 154.78, 171.04; HRMS (ESI-TOF) calculated for C26H34N2O4Na [M + Na]+: 461.2416; found: 461.2419.(S)-3-((2R,3R)-2-(Benzyloxy)-3-hydroxypentanoyl)-4-isopropyl-1-((R)-1-phenylethyl)-imidazolidin-2-one (3k)
Yield = 82%; gummy. [α]20D = +71.3 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.83 (d, J = 6.9 Hz, 3H), 0.88 (d, J = 7.0 Hz, 3H), 1.04 (t, J = 7.4 Hz, 3H), 1.54–1.63 (m, 1H), 1.57 (d, J = 7.2 Hz, 3H), 1.82–1.92 (m, 1H), 2.38–2.46 (m, 1H), 2.93 (t, J = 9.4 Hz, 1H), 3.11 (dd, J = 2.4, 9.7 Hz, 1H), 3.74–3.81 (m, 1H), 4.04–4.08 (m, 1H), 4.54 (dd, J = 11.5, 15.2 Hz, 2H), 5.23 (d, J = 7.2 Hz, 1H), 5.23 (q, J = 7.2 Hz, 1H), 7.18–7.22 (m, 3H), 7.32–7.45 (m, 7H); 13C NMR (100 MHz, CDCl3) δ 9.64, 14.48, 16.10, 17.96, 27.35, 28.72, 37.67, 50.61, 55.60, 72.89, 74.43, 79.75, 127.25, 127.80, 128.17, 128.22, 128.60, 128.82, 137.48, 138.81, 155.01, 171.95; HRMS (ESI-TOF) calculated for C26H34N2O4Na [M + Na]+: 461.2416; found: 461.2419.(S)-3-((2R,3R)-2-(Benzyloxy)-3-(4-((tert-butyldimethylsilyl)oxy)-3-methoxyphenyl)-3-hydroxypropanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3l)
Yield = 85%; gummy. [α]20D = +123.5 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.19 (d, J = 1.6 Hz, 6H), 0.77 (d, J = 6.9 Hz, 3H), 0.88 (d, J = 7.0 Hz, 3H), 1.04 (s, 9H), 1.59 (d, J = 7.2 Hz, 3H), 2.34–2.42 (m, 1H), 2.89 (t, J = 9.4 Hz, 1H), 3.08 (dd, J = 2.3, 9.7 Hz, 1H), 3.82 (s, 3H), 4.00 (d, J = 9.5 Hz, 1H), 4.02–4.05 (m, 1H), 4.38 (d, J = 1.9 Hz, 2H), 4.80 (t, J = 9.1 Hz, 1H), 5.35 (q, J = 7.2 Hz, 1H), 5.51 (d, J = 8.1 Hz, 1H), 6.88 (d, J = 8.0 Hz, 1H), 7.00–7.05 (m, 2H), 7.11–7.18 (m, 5H), 7.33–7.45 (m, 5H); 13C NMR (100 MHz, CDCl3) δ −4.61, −4.58, 14.49, 16.07, 18.00, 18.50, 25.78, 28.78, 37.62, 50.57, 55.43, 55.77, 73.01, 75.07, 79.50, 111.37, 118.96, 120.68, 127.25, 127.66, 128.06, 128.17, 128.45, 128.82, 135.51, 137.40, 138.76, 144.54, 150.61, 155.04, 171.83; HRMS (ESI-TOF) calculated for C37H50N2O4SiNa [M + Na]+: 669.3336; found: 669.3333.(S)-3-((2R,3R)-2-(Benzyloxy)-3-(4-((tert-butyldimethylsilyl)oxy)-3,5-dimethoxyphenyl)-3-hydroxypropanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3m)
Yield = 85%; gummy. [α]20D = +103.2 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.18 (d, J = 4.3 Hz, 6H), 0.75 (d, J = 6.9 Hz, 3H), 0.87 (d, J = 7.0, 3H), 1.06 (s, 9H), 1.57 (d, J = 7.2, 3H), 2.33–2.41 (m, 1H), 2.88 (t, J = 9.4 Hz, 1H), 3.07 (dd, J = 2.2, 9.7 Hz, 1H), 3.81 (s, 6H), 4.01–4.05 (m, 1H), 4.13 (d, J = 9.1 Hz, 1H), 4.39 (s, 2H), 4.82 (t, J = 8.6 Hz, 1H), 5.34 (q, J = 7.1, 1H), 5.54 (d, J = 8.0, 1H), 6.76 (s, 2H), 7.08–7.17 (m, 5H), 7.32–7.44 (m, 5H); 13C NMR (100 MHz, CDCl3) δ −4.60, −4.55, 14.47, 16.08, 17.99, 18.78, 25.90, 28.80, 37.59, 50.57, 55.70, 55.75, 73.01, 75.33, 79.29, 104.00, 127.23, 127.66, 128.05, 128.17, 128.43, 128.82, 133.70, 134.41, 138.78, 151.34, 154.94, 171.88; HRMS (ESI-TOF) calculated for C38H52N2O7SiNa [M + Na]+: 699.3441; found: 699.3448.(S)-3-((2R,3R)-2-(Benzyloxy)-3-hydroxy-4-methylpentanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3n)
Yield = 78%; gummy. [α]20D = +86.3 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.81 (d, J = 6.9 Hz, 3H), 0.86 (d, J = 7.0 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H), 1.03 (d, J = 7.0 Hz, 3H), 1.56 (d, J = 7.2 Hz, 3H), 2.10–2.18 (m, 1H), 2.38–2.49 (m, 1H), 2.88 (t, J = 9.4 Hz, 1H), 3.07 (dd, J = 2.3, 9.7 Hz, 1H), 3.16 (d, J = 11.0 Hz, 1H), 3.62–3.68 (m, 1H), 3.99–4.03 (m, 1H), 4.50 (dd, J = 11.6 Hz, 13.6, 2H), 5.23 (d, J = 9.0 Hz, 1H), 5.32 (q, J = 7.1 Hz, 1H), 7.14–7.20 (m, 2H), 7.28–7.45 (m, 8H); 13C NMR (100 MHz, CDCl3) δ 14.54, 16.01, 17.95, 19.82, 28.77, 29.70, 37.72, 50.54, 55.80, 72.75, 77.16, 127.16, 127.29, 127.86, 128.22, 128.76, 128.81, 137.33, 138.74, 155.41, 172.78; HRMS (ESI-TOF) calculated for C27H36N2O4Na [M + Na]+: 475.2573; found: 475.2557.Synthesis of (1R,2S)-1-(4-hydroxy-3-methoxyphenyl)propane-1,2,3-triol (4)
To a stirring solution of (S)-3-((2R,3R)-2-(benzyloxy)-3-(4-((tert-butyldimethylsilyl)oxy)-3-methoxyphenyl)-3-hydroxypropanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one 3l (1.0 g, 1.5 mmol, 1 eq.) in MeOH (25) mL was added NaBH4 (0.58 g, 15.5 mmol, 10 equiv.) and stirred for 3 h. The progress of the reaction was monitored by TLC. After the completion of the reaction, the solvent was evaporated under vacuum. The reaction mixture was extracted with EtOAc and the combined organic layers were dried over anhydrous sodium sulfate and concentrated in vacuum. The mixture was taken to the next step without purification.The above product was dissolved in THF (20 mL) and TBAF (1.55 mL, 1 M solution in THF) was added at 0 °C under nitrogen atmosphere and stirred for 30 min at rt until the complete consumption of starting material was observed in TLC. The mixture was extracted with EtOAc and the combined organic layers were dried over anhydrous sodium sulfate and concentrated in vacuum. The reaction mixture was then taken in MeOH and added to which Pd/C was added. It was then degassed and subjected to hydrogenolysis at a pressure of 5 atm for 3 h on a parr hydrogenator. After completion of the reaction, the reaction mixture was filtered through Celite-545 pad and it was purified by column chromatography on silica gel (60–120 mesh) using ethyl acetate–hexane (04:06) mixture as eluent.
Yield = 75%; white solid, mp = 80 °C [found [α]20D = +9.8 (c 0.8 in CHCl3), lit. [α]D = +9.4 (c 0.8)];7 1H NMR (400 MHz, MeOD) δ 3.51 (dd, J = 6.6, 11.3 Hz, 1H), 3.60 (dd, J = 3.7, 11.3 Hz, 1H), 3.65–3.69 (m, 1H), 3.78 (s, 3H), 4.48 (d, J = 6.2 Hz, 1H), 6.70 (d, J = 8.1 Hz, 1H), 6.75 (dd, J = 1.7, 8.1 Hz, 1H), 6.93 (d, J = 1.6 Hz, 1H); 13C NMR (100 MHz, MeOD) δ 47.12, 47.34, 47.55, 47.76, 47.98, 48.19, 48.40, 55.04, 62.64, 74.64, 75.24, 110.32, 114.50, 119.50, 133.36, 145.38, 147.34; HRMS (ESI-TOF) calculated for C10H14O5Na [M + Na]+: 237.0739; found: 237.0734.
Synthesis of (2R,3R)-2-(benzyloxy)-3-hydroxy-3-phenyl propanoic acid (5)
To a stirring solution of (S)-3-((2R,3R)-2-(benzyloxy)-3-hydroxy-3-phenylpropanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one 3a (0.2 g, 0.4 mmol, 1 equiv.) in THF:H2O (3:1) was added in H2O2 (0.46 mL, 4.1 mmol, 10 equiv., 30% solution v/v in water) and LiOH·H2O (0.03 g, 0.8 mmol, 2 equiv.) at 0 °C and stir for 1 h at rt. The reaction was then quenched with 10% excess of 1.5 N Na2SO3 solution. Sodium bicarbonate was added to the reaction mixture to get the pH at 9–10. THF was evaporated and the reaction mixture was extracted with DCM to recover the chiral auxiliary. The aqueous layer was acidified to pH 1–2 using concentrated HCl and extracted with DCM, washed with water, brine dried over anhydrous sodium sulfate and concentrated to get the pure product.
Yield = 85%; colorless oil. [α]20D = +34.7 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.14 (d, J = 6.4 Hz, 1H), 4.36 (d, J = 11.6 Hz, 1H), 4.64 (d, J = 11.6 Hz, 1H), 5.01 (d, J = 6.4 Hz, 1H), 5.41 (brs, 2H), 7.12–7.14 (m, 2H), 7.27–7.39 (m, 8H); 13C NMR (100 MHz, CDCl3) δ 73.28, 74.24, 81.57, 126.95, 128.14, 128.28, 128.34, 128.47, 128.62, 136.46, 139.25, 174.17; HRMS (ESI-TOF) calculated for C16H16O4Na [M + Na]+: 295.0945; found: 295.0944.
Synthesis of (2R,3R)-ethyl-2-(benzyloxy)-3-hydroxy-3-phenylpropanoate (6)
To a stirring solution of (S)-3-((2R,3R)-2-(benzyloxy)-3-hydroxy-3-phenylpropanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one 3a (0.2 g, 0.4 mmol, 1 equiv.) in EtOH 5 mL was added K2CO3 (0.57 g, 4.1 mmol, 10 equiv.) and stirred for 4 h. The progress of the reaction was monitored by TLC. The reaction mixture was filtered and solvent was evaporated under vacuum. The crude reaction mixture was purified by column chromatography using silica gel (60–120 mesh) eluting with petroleum ether/EtOAc (09:01) to afford the desired product.
Yield = 90%; colorless oil. [α]20D = +36.7 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.19 (t, J = 7.1 Hz, 3H), 4.14 (d, J = 6.0 Hz, 1H), 4.16 (q, J = 7.2 Hz, 2H), 4.40 (d, J = 11.6 Hz, 1H), 4.70 (d, J = 11.6 Hz, 1H), 5.03 (d, J = 6.1 Hz, 1H), 7.20–7.23 (m, 2H), 7.30–7.41 (m, 8H); 13C NMR (100 MHz, CDCl3) δ 14.06, 61.11, 72.96, 74.20, 81.85, 126.81, 128.00, 128.05, 128.08, 128.20, 128.41, 136.90, 139.61, 170.66; HRMS (ESI-TOF) calculated for C18H20O4Na [M + Na]+: 323.1259; found: 323.1278.
Synthesis of (1R,2S)-2-(benzyloxy)-1-phenylpropane-1,3-diol (7)
To a stirred solution of (S)-3-((2R,3R)-2-(benzyloxy)-3-hydroxy-3-phenylpropanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one 3a (0.2 g, 0.4 mmol, 1 equiv.) in MeOH 5 mL was added NaBH4 (0.15 g, 4.1 mmol, 10 equiv.) and stirred for 3 h. The progress of the reaction was monitored by TLC. The solvent was evaporated under vacuum. The crude was purified by column chromatography using silica gel (60–120 mesh) by eluting with petroleum ether/EtOAc (7:3) to afford the desired product.
Yield = 88%; colorless oil. [α]20D = +48.0 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 2.85 (brs, 2H), 3.59 (dd, J = 4.6, 5.2 Hz, 1H), 3.75 (dq, J = 4.7, 11.9 Hz, 2H), 4.44 (d, J = 11.5 Hz, 1H), 4.52 (d, J = 11.5 Hz, 1H), 4.92 (d, J = 5.8 Hz, 1H), 7.21–7.39 (m, 10H); 13C NMR (100 MHz, CDCl3) δ 61.45, 72.32, 74.17, 82.33, 126.45, 127.77, 127.94, 128.01, 128.40, 128.49, 137.73, 141.04; HRMS (ESI-TOF) calculated for C16H18O3Na [M + Na]+: 281.1154; found: 281.1098.
Synthesis of (2R,3R)-N-benzyl-2-(benzyloxy)-3-hydroxy-3-phenylpropanamide (8)
To a stirred solution of (S)-3-((2R,3R)-2-(benzyloxy)-3-hydroxy-3-phenylpropanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one 3a (0.2 g, 0.4 mmol, 1 equiv.) in THF (10 mL) was added benzylamine (0.06 mL, 0.6 mmol, 1.5 equiv.) and stirred for 30 min. The progress of the reaction was monitored by TLC. The reaction mixture was extracted with EtOAc and the combined organic layers were dried over anhydrous sodium sulfate and concentrated in vacuum. The crude product was purified by flash column chromatography using silica gel (60–120 mesh) by eluting with petroleum ether/EtOAc (6:4) to afford the desired product.
Yield = 78%; sticky. [α]20D = +30.4 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.03 (d, J = 7.3 Hz, 1H), 4.21 (q, J = 11.3 Hz, 2H), 4.42 (d, J = 6.0 Hz, 2H), 4.91 (d, J = 7.3 Hz, 1H), 7.01 (bs, 1H), 7.05–7.08 (m, 2H), 7.12–7.14 (m, 2H), 7.26–7.29 (m, 3H), 7.30–7.34 (m, 3H), 7.35–7.39 (m, 3H), 7.47–7.50 (m, 2H); 13C NMR (100 MHz, CDCl3) 42.94, 74.51, 75.28, 81.93, 127.46, 127.57, 127.61, 128.22, 128.27, 128.31, 128.37, 128.63, 128.74, 136.44, 137.43, 139.88, 171.74; HRMS (ESI-TOF) calculated for C22H23NO3Na [M + Na]+: 384.1576; found: 384.1576.
4-((tert-Butyldimethylsilyl)oxy)-3,5-dimethoxybenzaldehyde (9)
To a stirring solution of syringaldeyhde (2.0 g, 11.0 mmol, 1 equiv.) in DCM (30 mL) at 0 °C was added imidazole (1.87 g, 27.4 mmol, 2.5 equiv.) and stirred for 30 min. TBSCl (1.82 g, 12.1 mmol, 1.1 eq.) was then added to the reaction mixture at 0 °C and stirred for another 1 h at rt. The progress of the reaction was monitored by TLC. The reaction mixture was extracted with DCM and the combined organic layers were dried over anhydrous sodium sulfate and concentrated in vacuum. The crude reaction mixture was purified by column chromatography using silica gel (60–120 mesh) by eluting with petroleum ether/EtOAc (97:03) to afford the desired product.
Off white solid, yield = 92%, mp = 75–76 °C; [lit. mp = 78–79 °C];8 1H NMR (400 MHz, CDCl3) δ 0.17 (s, 6H), 1.03 (s, 9H), 3.88 (s, 6H), 7.11 (s, 2H), 9.83 (s, 1H); 13C NMR (100 MHz, CDCl3) δ −4.55, 18.77, 25.66, 55.78, 106.71, 129.36, 140.66, 151.97, 191.00.
(E)-Ethyl-3-(4-hydroxy-3,5-dimethoxyphenyl)acrylate (10)
To 4-((tert-butyldimethylsilyl)oxy)-3,5-dimethoxybenzaldehyde (9) (4.08 g, 13.49 mmol, 1.0 equiv.) was added DBU (2.11 mL, 14.2 mmol, 1.05 equiv.) and triethyl phosphonoacetate (2.81 mL, 14.16 mmol, 1.05 equiv.) and stirred for 4 h. After completion the reaction mixture was extracted with the ethyl acetate. The organic layer was washed with 1 N HCl solution and dried over sodium sulfate to give the crude product. The crude product without purification was taken in acetonitrile–water mixture (40 mL, 38:02) and DBU (2.32 mL, 15.57 mL) was added and stirred for 3 h. The reaction mixture was extracted with ethyl acetate. Organic layer was washed with 1 N HCl, water and dried over anhydrous sodium sulfate. The solvent was concentrated in vacuum. The crude reaction mixture was purified by column chromatography using silica gel (60–120 mesh) by eluting with petroleum ether/EtOAc (97:03) to afford the desired product.
Light yellow solid, yield = 90%, mp; experimental = 82 °C; lit. = 83 °C; 1H NMR (400 MHz, CDCl3) δ 1.35 (t, J = 7.1 Hz, 3H), 3.93 (s, 6H), 4.27 (q, J = 7.1 Hz, 2H), 5.86 (brs, 1H), 6.32 (d, J = 15.9 Hz, 1H), 6.78 (s, 2H), 7.61 (d, J = 15.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.35, 25.65, 56.32, 60.42, 105.00, 116.00, 125.93, 137.07, 144.89, 147.21, 167.19.
(E)-Ethyl-3-(4-hydroxy-3-methoxyphenyl)acrylate (11)
To the stirred solution of ferulic acid (2.0 g, 8.92 mmol, 1.0 equiv.) in EtOH was added catalytic amount of H2SO4 and refluxed for 4 h. After completion of the reaction the solvent was removed under vacuum and the compound was extracted with ethyl acetate. Organic layer was washed with aq. NaHCO3 solution, water and dried over anhydrous sodium sulfate. The solvent was evaporated under vacuum to give the pure product (2.11 g, 94%).Light reddish solid; yield = 94%, mp; experimental = 66 °C, lit. = 63–65 °C; 1H NMR (400 MHz, CDCl3) δ 1.34 (t, J = 7.2 Hz, 3H), 3.91 (s, 3H), 4.27 (q, J = 7.1 Hz, 2H), 6.16 (s, 1H), 6.30 (d, J = 15.9 Hz, 1H), 6.92 (d, J = 8.2 Hz, 1H), 7.03 (d, J = 1.8 Hz, 1H), 7.07 (dd, J = 1.8, 8.2 Hz, 1H), 7.62 (d, J = 15.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.33, 55.90, 60.39, 109.47, 114.83, 115.53, 122.98, 126.97, 144.76, 146.89, 148.04, 167.40.
(S)-3-(2-Bromoacetyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (12)
To a stirred solution of (S)-4-isopropyl-1-[(R)-1-phenylethyl]imidazolidin-2-one (8 g, 34.4 mmol, 1.0 equiv.) in dry THF (100 mL) and under N2 environment was added sodium hydride (60% suspension in paraffin oil, 1.7 g, 41.3 mmol, 1.2 equiv.) in portion-wise at 0 °C. After 1 h, bromoacetyl bromide (3.3 mL, 37.93 mmol, 1.1 equiv.) was added drop-wise and stirring was continued for another hour. The reaction mixture was quenched with aq. NH4Cl, washed with aq. NaHCO3 and dried over anhydrous sodium sulfate. It was evaporated under vacuum to give a gummy compound.Yield = 80%; yellowish liquid. [α]20D = +67.7 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.87 (d, J = 6.9 Hz, 3H), 0.91 (d, J = 7.0 Hz, 3H), 1.62 (d, J = 7.2 Hz, 3H), 2.40–2.48 (m, 1H), 2.99 (t, J = 9.5 Hz, 1H), 3.14 (dd, J = 2.9, 9.6 Hz, 1H), 4.23 (dt, J = 3.3, 9.4 Hz, 1H), 4.53 (d, J = 11.8 Hz, 1H), 4.74 (d, J = 11.8 Hz, 1H), 5.39 (q, J = 7.2 Hz, 1H), 7.32–7.42 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 14.44, 16.06, 17.98, 28.44, 29.08, 37.34, 50.53, 55.51, 127.13, 128.06, 128.80, 138.76, 153.84, 166.34; HRMS (ESI-TOF) calculated for C16H21BrN2O2Na [M + Na]+: 375.0684; found: 375.0668.
(E)-4-(3-Hydroxyprop-1-en-1-yl)-2-methoxyphenol (13a)
To a stirred suspension of LiAlH4 (2.56 g, 67.36 mmol) in freshly dried THF (200 mL), a solution of BnCl (7.7 mL, 67.36 mmol) in dry THF (20 mL) was added dropwise through dropping funnel at 0 °C. After the addition of BnCl the solution was allowed to warm to room temperature and stirred for 15–20 min at room temperature. To this, a solution of (E)-ethyl-3-(4-hydroxy-3-methoxyphenyl) acrylate (10 g, 45 mmol) in THF (40 mL) was added dropwise. The reaction mixture was allowed to stir at room temperature for 2 h. Thereafter it was cooled and slowly quenched with water, aq. NaOH and again washed with water to form slurry which was filtered through celite bed. The filtrate was taken in ethyl acetate, washed with brine, and dried over anhydrous sodium sulfate. It was evaporated under vacuum to afford the desired product.9Light yellow solid; yield = 85%, mp; experimental = 74 °C, lit. = 75–80 °C; 1H NMR (400 MHz, CDCl3) δ 3.92 (s, 3H), 4.32 (dd, J = 1.2, 6.0 Hz, 2H), 5.78 (s, 1H), 6.24 (dt, J = 6.0, 15.8 Hz, 1H), 6.55 (d, J = 15.8 Hz, 1H), 6.87–6.94 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 55.88, 63.87, 108.33, 114.49, 120.31, 126.13, 129.24, 131.39, 145.58, 146.65.
(E)-4-(3-Hydroxyprop-1-en-1-yl)-2,6-dimethoxyphenol (13b)
Light yellow solid, yield = 84%, mp; experimental = 64 °C, lit. = 61–65 °C; 1H NMR (400 MHz, CDCl3) δ 3.91 (s, 6H), 4.33 (d, J = 5.9 Hz, 2H), 5.63 (bs, 1H), 6.26 (dt, J = 5.9, 15.7 Hz, 1H), 6.53 (d, J = 15.8 Hz, 1H), 6.65 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 56.27, 63.77, 103.29, 126.56, 128.21, 131.50, 134.73, 147.11.(S)-3-(2-(4-((E)-3-((tert-Butyldimethylsilyl)oxy)prop-1-en-1-yl)-2-methoxyphenoxy)acetyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (14a)
To a stirred solution of (E)-4-(3-hydroxyprop-1-en-1-yl)-2-methoxyphenol (2 g, 11.10 mmol) in 25 mL acetone at room temperature was added K2CO3 (3.83 g, 27.74 mmol) and KI (0.18 g, 1.109 mmol) and stirred for 15 minutes. A solution of (S)-3-(2-bromoacetyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (4.31 g, 12.20 mmol) in acetone (10 mL) was added to the reaction mixture. The reaction mixture was allowed to stir overnight at room temperature. After the completion of the reaction the solvent was evaporated and water was added and the organic layer was separated, washed with aq. NaHCO3 and brine. The solvent was evaporated under vacuum to give the crude product as a sticky liquid (4.77 g, 95%).To a solution of (S)-3-(2-(4-((E)-3-hydroxyprop-1-en-1-yl)-2-methoxyphenoxy)acetyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (3 g, 6.63 mmol) in DCM (40 mL) was added imidazole (1.13 g, 16.57 mmol) at 0 °C and stirred for 30 min. Then the solution of TBSCl (1 g, 6.63 mmol) in DCM (10 mL) was added to the reaction mixture. The reaction mixture was allowed to warm to room temperature and stirred for another 2 h. After the completion of the reaction water was added to the reaction mixture, the organic phase was separated and dried over anhydrous sodium sulfate. Solvent was evaporated under vacuum and the crude product was purified by column chromatography to give the product as a sticky liquid.
Yield = 89%; sticky. [α]20D = +137.7 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.13 (s, 6H), 0.85 (d, J = 6.9 Hz, 3H), 0.89 (d, J = 7.0 Hz, 3H), 0.96 (s, 9H), 1.61 (d, J = 7.2 Hz, 3H), 2.46–2.53 (m, 1H), 3.02 (t, J = 9.6 Hz, 1H), 3.16 (dd, J = 9.7, 3.02 Hz, 1H), 3.75–3.79 (m, 1H), 3.93 (s, 3H), 4.23 (dt, J = 9.5, 3.3 Hz, 1H), 4.35 (dd, J = 5.2, 1.6 Hz, 1H), 5.36 (q, J = 7.2 Hz, 1H), 5.38 (d, J = 2.3, 2H), 6.17 (dt, J = 15.7, 5.2 Hz, 1H), 6.52 (d, J = 15.8 Hz, 1H), 6.80 (d, J = 8.3 Hz, 1H), 6.88 (dd, J = 8.3, 1.8 Hz, 1H), 6.96 (d, J = 1.8 Hz, 1H), 7.33–7.42 (m, 5H). 13C NMR (100 MHz, CDCl3) δ −5.03, 14.39, 16.23, 18.02, 18.56, 26.08, 28.37, 37.95, 50.52, 55.01, 56.00, 64.10, 68.52, 109.68, 113.68, 119.29, 127.25, 127.56, 128.14, 128.89, 129.47, 131.24, 138.85, 147.31, 149.53, 154.55, 168.48; ESI-HRMS (m/z): [M + Na]+ calculated for C32H46N2SiNaO5, 589.3074, found, 589.3075.
(S)-3-(2-(4-((E)-3-((tert-Butyldimethylsilyl)oxy)prop-1-en-1-yl)-2,6-dimethoxypheno-xy)acetyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (14b)
Yield = 85%; sticky. [α]20D = +89.7 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.13 (6 s, 6H), 0.84 (d, J = 6.8 Hz, 3H), 0.89 (d, J = 7.0 Hz, 3H), 0.96 (s, 9H), 1.57 (d, J = 7.2 Hz, 3H), 2.46–2.54 (m, 1H), 2.98 (t, J = 9.5 Hz, 1H), 3.12 (dd, J = 2.8, 9.6 Hz, 1H), 3.84 (s, 6H), 4.25 (dt, J = 3.2 Hz, 1H), 4.35 (dd, J = 1.3, 5.0 Hz, 2H), 5.22–5.33 (m, 3H), 6.20 (dt, J = 5.1, 15.7 Hz, 1H), 6.51 (d, 15.8 Hz, 1H), 6.59 (s, 2H), 7.28–7.38 (m, 5H); 13C NMR (100 MHz, CDCl3) δ −5.12, 14.45, 16.10, 18.01, 18.48, 26.02, 28.49, 37.84, 50.27, 54.80, 56.19, 63.86, 71.92, 103.69, 127.12, 127.95, 128.53, 128.73, 129.56, 132.82, 136.36, 138.94, 152.93, 154.50, 169.08; ESI-HRMS (m/z): [M + Na]+ calculated for C33H48N2SiNaO6, 619.3175, found, 619.3180.(S)-3-((2R,3R)-2-(4-((E)-3-((tert-Butyldimethylsilyl)oxy)prop-1-en-1-yl)-2-methoxyph-enoxy)-3-(3,4-dimethoxyphenyl)-3-hydroxypropanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (15a)
To a solution of (S)-3-(2-(4-((E)-3-((tert-butyldimethylsilyl) oxy)prop-1-en-1-yl)-2-methoxyphenoxy)acetyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (1.0 g, 2.2 mmol, 1.0 equiv.) in anhydrous THF (5 mL) under N2 environment was added LiHMDS solution (2.4 mL, 2.4 mmol, 1.1 equiv.; 1 M in THF) at −78 °C, and stirred for 1 h. 3,4-Dimethoxybenzaldehyde (0.32 g, 2.4 mmol, 1.1 equiv.) dissolved in THF (3 mL) was then added into the reaction mixture and stirred for another hour. The reaction was quenched with saturated aq. NH4Cl solution, extracted with EtOAc and further washed with brine. It was dried over anhydrous sodium sulfate, concentrated and purified by column chromatography silica gel (60–120 mesh) by eluting with petroleum ether:EtOAc (6:4) to afford the desired product.
Yield = 78%; sticky. [α]20D = +115.7 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) 0.12 (s, 6H), 0.77 (d, J = 6.8 Hz, 3H), 0.89 (d, J = 7.0 Hz, 3H), 0.95 (s, 9H), 1.61 (d, J = 7.2 Hz, 3H), 2.32–2.40 (m, 1H), 2.99 (t, J = 9.5 Hz, 1H), 3.13 (dd, J = 2.3, 9.8 Hz, 1H), 3.67 (s, 3H), 3.88 (s, 3H), 3.92 (s, 3H), 4.26–4.29 (m, 1H), 4.33 (dd, J = 4.1, 5.2 Hz, 2H), 5.07 (brs, 1H), 5.38 (q, J = 7.0 Hz, 1H), 6.13 (dt, J = 5.2, 15.8 Hz, 1H), 6.26 (d, J = 7.5 Hz, 1H), 6.47 (d, J = 15.8 Hz, 1H), 6.56 (d, J = 8.2 Hz, 1H), 6.71 (dd, J = 1.8, 8.2 Hz, 1H), 6.84 (d, J = 1.8 Hz, 1H), 6.86 (d, J = 8.3 Hz, 1H), 7.13 (dd, J = 1.8, 8.3 Hz, 1H), 7.17 (d, J = 1.8 Hz, 1H), 7.31–7.41 (m, 6H); 13C NMR (100 MHz, CDCl3), −5.13, 14.51, 16.00, 17.98, 18.46, 24.69, 25.99, 37.74, 50.55, 55.61, 55.81, 55.85, 55.91, 63.89, 74.84, 79.91, 110.33, 110.74, 110.90, 118.08, 119.18, 119.30, 127.09, 128.01, 128.08, 128.47, 128.81, 129.12, 132.32, 133.73, 138.80, 146.77, 148.63, 150.42, 155.07, 170.35; ESI-HRMS (m/z): [M + Na]+ calculated for C42H58N2SiNaO9, 755.3704, found, 755.3703.
(S)-3-((2R,3R)-2-(4-((E)-3-((tert-Butyldimethylsilyl)oxy)prop-1-en-1-yl)-2-methoxyphenoxy)-3-(4-((tert-butyldiphenylsilyl)oxy)-3-methoxyphenyl)-3-hydroxypropanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (15b)
Yield = 75%; sticky. [α]20D = +98.7 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1H NMR (400 MHz, CDCl3) δ 0.14 (s, 6H), 0.79 (d, J = 6.8 Hz, 3H), 0.89 (d, J = 7.0 Hz, 3H), 0.97 (s, 9H), 1.13 (s, 9H), 1.61 (d, J = 7.1 Hz, 3H), 2.36–2.40 (m, 1H), 3.00 (t, J = 9.5, 1H), 3.14 (dd, J = 2.3, 9.8, 1H), 3.61 (s, 6H), 3.92 (d, J = 9.1 Hz, 1H), 4.26–4.30 (m, 1H), 4.35 (dd, J = 1.6, 5.2 Hz, 2H), 4.96 (t, J = 8.1 Hz, 1H), 5.37 (q, J = 7.1 Hz, 3H), 6.11–6.18 (m, 2H), 6.47–6.50 (m, 2H), 6.68 (dd, J = 1.9, 8.3 Hz, 1H), 6.70 (d, J = 8.3 Hz, 1H), 6.83 (d, J = 1.9 Hz, 1H), 6.88 (dd, J = 2.0, 8.3 Hz, 1H), 7.05 (d, J = 2.0 Hz, 1H), 7.3–7.43 (m, 11H), 7.71–7.75 (m, 4H); 13C NMR (100 MHz, CDCl3), −5.08, 1.06, 14.58, 16.00, 18.01, 18.50, 19.83, 20.69, 26.03, 26.74, 28.98, 29.72, 37.78, 50.56, 55.43, 55.68, 55.77, 63.95, 75.09, 80.18, 110.34, 112.02, 115.43, 118.35, 119.07, 119.35, 119.81, 120.08, 127.14, 127.50, 127.97, 128.11, 128.82, 129.22, 129.48, 129.57, 132.29, 133.72, 133.76, 134.48, 135.37, 138.80, 144.74, 146.94, 150.15, 150.42, 155.14, 170.64; ESI-HRMS (m/z): [M + Na]+ calculated for C56H72N2Si2NaO8, 979.4725, found, 979.4728.(S)-3-((2R,3R)-2-(4-((E)-3-((tert-Butyldimethylsilyl)oxy)prop-1-en-1-yl)-2,6-dimethoxyphenoxy)-3-(4-((tert-butyldiphenylsilyl)oxy)-3-methoxyphenyl)-3-hydroxy propanoyl)-4-isopropyl-1-((R)-1-phenylethyl)imidazolidin-2-one (15c)
Yield = 74%; sticky. [α]20D = +77.8 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.15 (s, 6H), 0.61 (d, J = 6.8 Hz, 3H), 0.83 (d, J = 7.0 Hz, 3H), 0.98 (s, 9H), 1.11 (s, 9H), 1.56 (d, J = 7.2 Hz, 3H), 2.11–2.16 (m, 1H), 2.96 (t, J = 9.5 Hz, 1H), 3.11 (dd, J = 2.2, 9.6 Hz, 1H), 3.59 (s, 3H), 3.60 (s, 6H), 4.28 (dt, J = 2.9, 9.5 Hz, 1H), 4.37 (dd, J = 1.5, 5.1 Hz, 2H), 4.44 (d, J = 8.8 Hz, 1H), 5.07 (t, J = 6.7 Hz, 1H), 5.29 (q, J = 7.1 Hz, 1H), 6.18 (d, J = 5.5 Hz, 1H), 6.20 (dt, J = 5.1, 15.8 Hz, 1H), 6.49–6.53 (m, 3H), 6.66 (d, J = 8.2 Hz, 1H), 6.81 (dd, J = 1.9, 8.2 Hz, 1H), 7.06 (d, J = 1.9 Hz, 1H), 7.27–7.30 (m, 2H), 7.32–7.42 (m, 9H), 7.7–7.75 (m, 4H); 13C NMR (100 MHz, CDCl3) δ −5.12, 14.29, 16.02, 18.08, 18.51, 19.82, 26.03, 26.71, 28.89, 29.72, 37.29, 50.36, 55.08, 55.31, 55.81, 63.83, 74.37, 82.54, 103.39, 111.79, 119.43, 127.16, 127.46, 127.47, 127.97, 128.64, 128.70, 129.44, 129.48, 129.50, 132.63, 133.38, 133.78, 133.90, 135.35, 135.38, 136.85, 139.11, 144.49, 150.00, 152.20, 154.25, 169.51; ESI-HRMS (m/z): [M + Na]+ calculated for C57H74N2Si2NaO9, 1009.4831, found, 1009.4833.(1R,2S)-1-(3,4-Dimethoxyphenyl)-2-(4-((E)-3-hydroxyprop-1-en-1-yl)-2-methoxyphenoxy)propane-1,3-diol (16a)
Yield = 80%; yellowish oil. [α]20D = +7.8 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 3.68 (dd, J = 3.4, 12.2 Hz, 1H), 3.87 (s, 3H), 3.88 (s, 3H), 3.90 (s, 3H), 3.92 (dd, J = 5.8, 12.2 Hz, 1H), 4.17 (m, 1H), 4.32 (dd, J = 1.5, 5.7 Hz, 2H), 4.99 (d, J = 4.8 Hz, 1H), 6.28 (dt, J = 5.7, 15.8 Hz, 1H), 6.56 (d, J = 15.8 Hz, 1H), 6.84 (d, J = 8.3 Hz, 1H), 6.88–6.92 (m, 3H), 6.97 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 55.94, 60.80, 63.62, 72.76, 87.30, 109.22, 109.85, 111.05, 118.41, 120.08, 120.71, 128.12, 130.52, 132.40, 133.04, 146.58, 148.53, 149.06, 151.52; ESI-HRMS (m/z): [M + Na]+ calculated for C21H26NaO7, 413.1576, found, 413.1582.(1R,2S)-1-(4-Hydroxy-3-methoxyphenyl)-2-(4-((E)-3-hydroxyprop-1-en-1-yl)-2-methoxyphenoxy)propane-1,3-diol (16b)
Yield = 82%; colourless oil. [α]20D = +10.8 (c 1, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 3.59 (m, 2H), 3.72 (s, 3H), 3.73 (s, 3H), 4.08 (m, 2H), 4.30 (m, 1H), 4.61 (t, J = 5.6 Hz, 1H), 4.7 (t, J = 4.86 Hz, 1H), 4.80 (t, J = 5.5 Hz, 1H), 5.32 (d, J = 4.6 Hz, 1H), 6.22 (15.9, 5.3 Hz, 1H), 6.43 (bd, J = 15.9 Hz, 1H), 6.67 (d, J = 8.0 Hz, 1H), 6.77 (dd, J = 8.1, 1.7 Hz, 1H), 6.84 (dd, J = 8.3, 1.8 Hz, 1H), 6.92 (d, J = 8.4 Hz, 1H), 6.99 (m, 1H), 6.99 (m, 2H), 8.76 (s, 1H); 13C NMR (100 MHz, DMSO-d6) 55.31, 55.49, 60.00, 61.52, 71.51, 83.59, 109.75, 111.30, 114.45, 115.41, 118.93, 119.34, 128.37, 128.45, 129.91, 133.09, 145.32, 146.85, 147.45, 149.60; ESI-HRMS (m/z): [M + Na]+ calculated for C20H24NaO9, 399.1420, found, 399.1428.(1R,2S)-1-(4-Hydroxy-3-methoxyphenyl)-2-(4-((E)-3-hydroxyprop-1-en-1-yl)-2,6-dimethoxyphenoxy)propane-1,3-diol (16c)
Yield = 74%; colourless oil. [α]20D = +6.7 (c 1, CHCl3). 1H NMR (400 MHz, CDCl3) δ 3.51 (dd, J = 2.6, 12. Hz, 1H), 3.84 (s, 9H), 3.90 (dd, J = 7.3, 14.0 Hz, 1H), 4.12–4.15 (m, 1H), 4.30 (d, J = 5.4 Hz, 2H), 5.00 (d, J = 3.7 Hz, 1H), 6.29 (dt, J = 5.5, 15.8 Hz, 1H), 6.53 (d, J = 15.8 Hz, 1H), 6.63 (s, 2H), 6.74 (d, J = 8.2 Hz, 1H), 6.82–6.84 (m, 1H), 6.94 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 55.93, 56.10, 60.52, 63.24, 72.49, 87.03, 103.45, 108.56, 114.32, 118.77, 128.95, 130.37, 131.22, 133.48, 134.44, 144.90, 146.71, 153.27; ESI-HRMS (m/z): [M + Na]+ calculated for C21H28NaO8, 429.1525, found, 429.1520.(1R,2S)-1-(3,4-Dimethoxyphenyl)-2-(4-(3-hydroxypropyl)-2-methoxyphenoxy)propane-1,3-diol (17)
To a solution of (1R,2S)-1-(3,4-dimethoxyphenyl)-2-(4-((E)-3-hydroxyprop-1-en-1-yl)-2-methoxyphenoxy)propane-1,3-diol (16a) (0.1 g, 0.25 mmol) in ethyl (15 mL) acetate was added catalytic amount of Pd/C. It was then degassed and subjected to hydrogenolysis at a pressure of 5 atm for 1 h on a parr hydrogenator. After completion of the reaction, the reaction mixture was filtered through celite-545 pad and the filtrate thus obtained was concentrated in vacuum to get the pure product.Yield = 94%; colourless oil [reported [α]18D = 7.7 (c 1.9, MeOH), observed [α]20D = 9.2 (c 1.9, MeOH).10 1H NMR (400 MHz, CDCl3) 1.76 (m, 2H), 2.55 (t, J = 7.2 Hz, 2H), 3.55 (m, 2H), 3.64 (m, 1H), 3.74 (s, 3H), 3.77 (s, 3H), 3.77 (s, 3H), 3.82 (m, 1H), 4.07 (m, 1H), 4.90 (d, J = 3.5 Hz, 1H), 6.60 (d, J = 8.0 Hz, 1H), 6.66 (brs, 1H), 6.72 (d, J = 8.0 Hz, 1H), 6.75 (d, J = 8.0 Hz, 1H), 6.84 (d, J = 8.0, 1H), 6.92 (brs, 1H); 13C NMR (100 MHz, CDCl3) δ 31.5, 33.9, 55.6, 55.6, 55.6, 60.6, 61.6, 72.5, 86.3, 109.4, 110.8, 112.2, 118.4, 119.6, 120.9, 132.9, 137.4, 144.7, 148.1, 148.6, 150.6; HRMS (ESI-TOF) calculated for C21H28O7Na [M + Na]+: 415.1733; found: 415.1729.
Acknowledgements
Financial assistance in the form of a Senior Research Fellowship from CSIR to M. G. is gratefully acknowledged. We are thankful to Dr Angshuman Roy Choudhury, IISER Mohali for X-ray crystallographic data.References
- R. Mahrwald, in Modern Methods in Stereoselective Aldol Reactions, Wiley-VCH Verlag GmbH & Co. KGaA, 2013 Search PubMed.
- (a) K. S. Kim and S. D. Hong, Tetrahedron Lett., 2000, 41, 5909 CrossRef; (b) M. B. Andrus, B. V. S. Sekhar, T. M. Turner and E. L. Meredith, Tetrahedron Lett., 2001, 42, 7197 CrossRef CAS; (c) M. T. Crimmins and J. She, J. Am. Chem. Soc., 2004, 126, 12790 CrossRef CAS PubMed; (d) D. A. Evans, S. L. Bender and J. Morris, J. Am. Chem. Soc., 1988, 110, 2506 CrossRef CAS; (e) S. Fanjul and A. N. Hulme, J. Org. Chem., 2008, 73, 9788 CrossRef CAS PubMed; (f) D. A. Evans, J. R. Gage, J. L. Leighton and A. S. Kim, J. Org. Chem., 1992, 57, 1961 CrossRef CAS; (g) M. B. Andrus, B. V. S. Sekhar, E. L. Meredith and N. K. Dalley, Org. Lett., 2000, 2, 3035 CrossRef CAS PubMed; (h) M. T. Crimmins and P. J. McDougall, Org. Lett., 2003, 5, 591 CrossRef CAS PubMed; (i) W. R. Roush, L. A. Pfeifer and T. G. Marron, J. Org. Chem., 1998, 63, 2064 CrossRef CAS; (j) J. Bajget, M. Caba, E. Galvez, P. Romea, F. Urpi and M. F. Bardia, J. Org. Chem., 2012, 77, 8809 CrossRef PubMed; (k) M. T. Crimmins and A. L. Choy, J. Am. Chem. Soc., 1999, 121, 5653 CrossRef CAS; (l) M. Carda, J. Murga, E. Falomir, F. González and J. A. Marco, Tetrahedron, 2000, 56, 677 CrossRef CAS; (m) I. Paterson, D. J. Wallace and S. M. Velázquez, Tetrahedron Lett., 1994, 35, 9083 CrossRef CAS; (n) J. Murga, E. Falomir, F. González, M. Carda and J. A. Marco, Tetrahedron, 2002, 58, 9697 CrossRef CAS.
- Chiral catalysts and ligands for aldol reaction; (a) S. G. Nelson, Tetrahedron: Asymmetry, 1998, 9, 357 CrossRef CAS; (b) M. B. Andrus, J. Liu, Z. Ye and J. F. Cannon, Org. Lett., 2005, 7, 3861 CrossRef CAS PubMed; (c) E. M. Carreira, A. Fettes and C. Marti, Org. React., 2006, 67, 1 CAS; (d) C. Guillena, C. Najera and D. J. Ramon, Tetrahedron: Asymmetry, 2007, 18, 2249 CrossRef; (e) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600 RSC; (f) M. Markert, M. Mulzer, B. Schetter and R. Mahrwald, J. Am. Chem. Soc., 2007, 129, 7258 CrossRef CAS PubMed; (g) S. E. Denmark and W. J. Chung, J. Org. Chem., 2008, 73, 4582 CrossRef CAS PubMed; (h) B. M. Trost, W. M. Seganish, C. K. Chung and D. Amans, Chem.–Eur. J., 2012, 18, 2948 CrossRef CAS PubMed.
- G. Khatik, V. Kumar and V. A. Nair, Org. Lett., 2012, 14, 2442 CrossRef CAS PubMed.
- Lignan isolation: (a) F. Cutillo, B. D'Abrosca, M. Dellagreca, A. Fiorentino and A. Zarelli, J. Agric. Food Chem., 2003, 51, 6165 CrossRef CAS PubMed; (b) E. J. Jeong, J. H. Cho, S. H. Sung, S. Y. Kim and Y. C. Kim, Bioorg. Med. Chem. Lett., 2011, 21, 2283 CrossRef CAS PubMed; (c) X. X. Huang, C. C. Zhou, L. Z. Li, F. F. Li, L. L. Lou, D. M. Li, T. Ikejima, Y. Peng and S. J. Song, Bioorg. Med. Chem. Lett., 2013, 23, 5599 CrossRef CAS PubMed; (d) S. Li, K. Landquist and A. F. A. Wallis, Phytochemistry, 1998, 49, 2125 CrossRef CAS; (e) F. Li and X. W. Yang, Phytochemistry, 2008, 69, 765 CrossRef CAS PubMed; (f) W. Xu, H. Wang, G. X. Zhou and X. S. Yao, J. Asian Nat. Prod. Res., 2010, 12, 874 CrossRef CAS PubMed; (g) J. Lee, E. K. Seo, D. S. Jang, T. J. Ha and J. P. Kim, Chem. Pharm. Bull., 2009, 57, 298 CrossRef CAS PubMed; (h) Y. B. Liua, J. J. Qina, X. R. Chenga, J. X. Zhua, F. Zanga, H. Z. Jin and W. D. Zhang, Helv. Chim. Acta, 2011, 94, 1685 CrossRef; (i) R. Q. Mei, Y. H. Wang, G. H. Du, G. M. Liu, L. Zhang and Y. X. Cheng, J. Nat. Prod., 2009, 72, 621 CrossRef CAS PubMed; (j) H. H. Xiao, Y. Dai, M. S. Wong and X. S. Yao, Fitoterapia, 2014, 94, 29 CrossRef CAS PubMed.
- Lignan Synthesis: (a) K. Li and R. F. Helm, J. Chem. Soc., Perkin Trans. 1, 1996, 2425 RSC; (b) S. Hishiyama, Y. Otsuka, M. Nakamura, S. Ohara, S. Kajita, E. Masai and Y. Katayama, Tetrahedron Lett., 2012, 53, 842 CrossRef CAS; (c) C. E. Rye and D. Barker, Eur. J. Med. Chem., 2013, 60, 240 CrossRef CAS PubMed; (d) J. Buendia, J. Mottweiler and C. Bolm, Chem.–Eur. J., 2011, 17, 13877 CrossRef CAS PubMed; (e) C. Curti, F. Zanardi, L. Battistini, A. Sartori, G. Rassu, L. Pinna and G. Casiraghi, J. Org. Chem., 2006, 71, 8552 CrossRef CAS PubMed; (f) P. R. Reddy and B. Das, RSC Adv., 2014, 4, 7432 RSC; (g) S. K. Das, S. K. Das and G. Panda, Eur. J. Org. Chem., 2010, 5100 Search PubMed; (h) M. Nagaraju, R. Chandra, B. Bhimrao and B. B. Gawali, Synlett, 2012, 23, 1485 CrossRef CAS; (i) M. Chouhan, R. Sharma and V. A. Nair, Org. Lett., 2012, 14, 5672 CrossRef CAS PubMed.
- M. DellaGreca, A. Fiorentin, P. Monaco and L. Previtera, Synth. Commun., 1998, 28, 3693 CrossRef CAS.
- G. P. Pettit, N. Melody, A. Thornhill, J. C. Knight, T. L. Gory and C. L. Herald, J. Nat. Prod., 2009, 72, 1637 CrossRef CAS PubMed.
- X. Wang, X. Li, J. Xue, Y. Zhao and Y. Zhang, Tetrahedron Lett., 2009, 50, 413 CrossRef CAS.
- W. Xu, H. Wang, G. X. Zhou and X. S. Yao, J. Asian Nat. Prod. Res., 2010, 12, 874 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1452671. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra22026f |
| This journal is © The Royal Society of Chemistry 2016 |