
Potential roles of annexin A7 GTPase in autophagy, senescence and apoptosis
Abstract
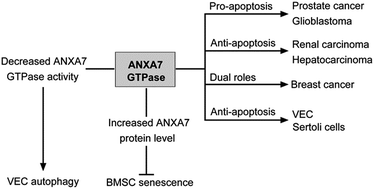
Annexin A7 (ANXA7) is a Ca2+-dependent phospholipid binding protein with intrinsic GTPase activity, belonging to the annexin superfamily. Accumulating research reveals that ANXA7 participates in cell autophagy, senescence and apoptosis in various cell types including vein endothelial cells (VECs), bone marrow-derived mesenchymal stem cells (BMSCs), Sertoli cells and cancer cells. This implies essential roles for ANXA7 in the physiological regulation of mammalian cells. Decreased ANXA7 GTPase activity induced by 6-amino-2,3-dihydro-3-hydroxymethyl-1,4-benzoxazine (ABO) promoted autophagy either by reducing the phosphorylation of ANXA7-interacting proteins or by inhibiting phosphatidylcholine-specific phospholipase C (PC-PLC) activity. ANXA7 inhibited senescence in BMSCs and protected cells from apoptosis in VECs and Sertoli cells. In cancer cells, ANXA7 had differing effects: it had a pro-apoptotic effect on prostate cancer and glioblastoma cells but an anti-apoptotic effect on renal cancer and hepatocarcinoma cells, and it played dual roles in breast cancer. The roles of ANXA7 GTPase activity in autophagy, senescence and apoptosis as well as its potential action mechanisms are summarized and discussed in the present review. An understanding of the global architecture of ANXA7 among autophagy, senescence and apoptosis interactive networks would be instrumental in providing novel therapeutic strategies for various human diseases.
 Please wait while we load your content...
Please wait while we load your content...

