DOI:
10.1039/C6RA21536J
(Paper)
RSC Adv., 2016,
6, 98221-98227
Thermally triggered optical tuning of π-conjugated graft copolymers based on reversible Diels–Alder reaction†
Received
27th August 2016
, Accepted 10th October 2016
First published on 14th October 2016
Abstract
In order to design a π-conjugated polymer film with tunable optical properties by thermally triggered activation of energy transfer after processing, two monodisperse phenylene ethynylene based oligomers with different optical properties were synthesized and attached to aliphatic polymers as π-conjugated side chains. Subsequently, the exchange of the side chain chromophores between the prepared donor and acceptor graft polymers in the solid state based on a reversible Diels–Alder reaction was studied in detail. The resulting donor–acceptor graft copolymer exhibits intra polymer energy transfer upon excitation of the donor moiety. The photophysical properties of the original and exchanged graft copolymers were investigated by means of absorption and emission spectroscopy. This novel concept opens the possibility for optical tuning of π-conjugated polymer films after processing as well as applications as thermally triggered sensor systems.
Introduction
During the last decade, graft polymers gained more and more attention in several application fields, e.g., photonics,1 thermo-responsive polymers,2 elastomers,3,4 nanostructures5,6 and bio-medicine.7,8 In graft polymers, side chains offer the possibility of dense packing, resulting in compact and confined structures.9 In order to combine the well-defined optoelectronic properties of monodisperse oligomer moieties and typical polymer properties, e.g., mechanical and chemical stability as well as processability into thin films, conjugated side chains have been attached to aliphatic polymers. Several π-conjugated oligomers have been successfully implemented as side chains, for instance triphenylamines,10–13 oligothiophenes,14 carbazoles,15 perylenes,16 phenylene ethynylenes17 and diphenyl acetylenes.18 The dense packing of side chain chromophores in such graft polymers by π–π-stacking resulted in a promising electronic communication between the π-conjugated moieties. In some cases, UV-vis absorption and emission spectroscopy has been utilized to further characterize these materials and to investigate energy transfer mechanisms between pendant acceptor and donor molecules.17,19,20 However, to the best of our knowledge, in all cases the final optical properties were introduced during the synthesis and before processing the polymers into films. Therefore, a polymer film for optoelectronic applications, in which energy transfer can be activated by thermal triggering resulting in changed optoelectronic properties, would represent a promising approach for optical tuning of a π-conjugated polymer film after processing.
For this purpose, the synthesis and design of novel graft copolymers is demonstrated, which combine the reversibility of the Diels–Alder reaction21–24 with the optical properties of π-conjugated oligomers. Therefore, one graft copolymer with rigid and planar π-conjugated oligomeric donor side chains as well as one graft copolymer with linear π-conjugated oligomeric acceptor side chains was prepared.25 Thermally triggered exchange by reversible Diels–Alder reaction between both different graft copolymers in the solid state resulted in a graft copolymer species with the same amount of donor and acceptor side chain chromophores. The exchanged graft copolymer exhibits an efficient energy transfer from the donor to the acceptor side chain, resulting in changed emissive properties.
Results and discussion
Monomer and oligomer synthesis
Two monodisperse conjugated phenylene ethynylene based oligomers (OPEs) were synthesized by multiple sequential Sonogashira cross-coupling reactions. The synthesis of the acceptor oligomeric side chain is depicted in ESI Scheme 1.† Several steps were carried out according to a literature procedure including small changes.17 Compound 3 was synthesized by a well-known procedure for alkylation of phenol moieties, followed by standard Sonogashira cross-coupling reaction with trimethylsilylacetylene (TMSA). Further Sonogashira cross-coupling reaction and deprotection steps resulted in compound 11. Subsequently, compound 10 was prepared by a simple condensation step. Another Sonogashira cross-coupling step with intermediate 11 resulted in the targeted conjugated acceptor oligomer with integrated Diels–Alder moiety and an extended π-conjugated system. In this manner, all compounds were synthesized in moderate up to quantitative yields. The synthesis of the targeted π-conjugated donor oligomer is schematically represented in ESI Scheme 2.† Mono bromination of compound 13 has been reached by slowly adding bromine at low temperatures to the reaction mixture. After further functionalization with iodine, intermediate 15 was selectively single cross-coupled via Sonogashira reaction with TMSA. Further Sonogashira cross-coupling and deprotection steps resulted in the targeted phenylene ethynylene based donor chromophore 19. Finally, the prepared donor oligomer 19 and acceptor oligomer 12 were coupled with furfuryl methacrylate (FMA), which is depicted in Scheme 1. In this manner, a reversible Diels–Alder function and a polymerizable group was introduced. Further details of the π-conjugated oligomer synthesis are summarized in the ESI.†
 |
| | Scheme 1 Schematic representation of introducing a polymerizable group in π-conjugated donor and acceptor oligomers. Reagents and conditions: (i) (a) chlorobenzene/120 °C, (b) FMA/CHCl3/55 °C. | |
Copolymer synthesis via “grafting to” method
Several “grafting to” methods based on Diels–Alder chemistry for graft,26 bottle brushed27 or comb copolymers28 were investigated in the last decades. Generally, the synthesis of the π-conjugated graft copolymers via “grafting to” method followed a two-step protocol. First, an aliphatic polymer backbone containing a diene had to be synthesized. Second, the side chains had to be attached to the polymer backbone via a Diels–Alder reaction. For this purpose, FMA and methyl methacrylate (MMA) as co-monomer were polymerized by standard procedure of atom transfer radical polymerization (ATRP) technique.24 Subsequently, the π-conjugated donor and acceptor oligomers were grafted to the polymer backbone via Diels–Alder reaction. MMA was introduced as comonomer, resulting in an increased distance between the rigid single oligomers within the final graft copolymer and, consequently, in a decreased steric hindrance. For this purpose, MMA, FMA, CuBr and 1,1,4,7,10,10-hexamethyltriethylene tetramine (HMTETA) as copper ligand were dissolved in toluene, whereby methyl α-bromoisobutyrate (MBiB) was utilized as a common initiator. For copolymer P1 a ratio of FMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MMA of 1:1 was utilized for polymerization. The molar mass and dispersity (Đ) values are summarized in Table 1. The resulting copolymer has a composition of 50% FMA and 50% MMA, which was calculated from the 1H NMR spectrum (ESI Fig. 1†). Separately, acceptor oligomer 12 was heated up to 120 °C in chlorobenzene to initiate the retro Diels–Alder reaction and, subsequently, to evaporate the released furan. The resulting maleimide functionalized acceptor oligomer was mixed with copolymer P1 in chlorobenzene. The solution was then drop-casted in order to obtain polymer films. The prepared films were heated up (55 °C) to initiate the Diels–Alder reaction, which finally resulted in acceptor containing graft copolymer P2 (Scheme 2). A similar procedure was carried out for the prepared donor dye 19, resulting in copolymer P3. Further details are summarized in ESI Scheme 3.† The molar mass and Đ values, determined by size exclusion chromatography (SEC), are summarized in Table 1.
:MMA of 1:1 was utilized for polymerization. The molar mass and dispersity (Đ) values are summarized in Table 1. The resulting copolymer has a composition of 50% FMA and 50% MMA, which was calculated from the 1H NMR spectrum (ESI Fig. 1†). Separately, acceptor oligomer 12 was heated up to 120 °C in chlorobenzene to initiate the retro Diels–Alder reaction and, subsequently, to evaporate the released furan. The resulting maleimide functionalized acceptor oligomer was mixed with copolymer P1 in chlorobenzene. The solution was then drop-casted in order to obtain polymer films. The prepared films were heated up (55 °C) to initiate the Diels–Alder reaction, which finally resulted in acceptor containing graft copolymer P2 (Scheme 2). A similar procedure was carried out for the prepared donor dye 19, resulting in copolymer P3. Further details are summarized in ESI Scheme 3.† The molar mass and Đ values, determined by size exclusion chromatography (SEC), are summarized in Table 1.
Table 1 Summarized molar mass and Đ values of prepared copolymers; adetermination by vapor pressure osmometry resulted in a Mn of 5353 g mol−1
| Copolymer |
M
n (g mol−1) |
M
w (g mol−1) |
Đ
|
|
P1
|
10000a |
13500 |
1.3 |
|
P2
|
23100 |
41300 |
1.8 |
|
P3
|
17100 |
26000 |
1.5 |
|
P4
|
9000 |
15400 |
1.6 |
|
P5
|
8100 |
12000 |
1.5 |
 |
| | Scheme 2 Schematic representation of the copolymer synthesis of P2via “grafting to” method. Reagents and conditions: (i) chlorobenzene/120 °C; (ii) copolymer P1/55 °C (drop casted film). | |
The composition of copolymers P2 and P3 with respect to the amounts of oligomer integrated as side chains and left free furan moieties has been studied by means of 1H NMR spectroscopy. The 1H NMR spectrum of copolymer P2 is depicted in Fig. 1. From the integrated signal of the CH2-groups next to the Diels–Alder based dye side chains (4.45 ppm, 4.75 ppm) and of the CH2-groups next to free furan groups (4.95 ppm), one can calculate an acceptor oligomer side chain content of approximately 36% and a free furan side chain content of 14%. The copolymer composition of copolymer P3 was estimated in the same manner, resulting in approximately 30% repeating units containing donor dyes and 20% free furan moieties (ESI Fig. 2†). The copolymer content of 50% MMA remained unchanged in the prepared graft copolymers P2 and P3 compared to copolymer P1. However, for efficient exchange studies of the oligomeric side chains between donor and acceptor graft copolymers, no free furan moieties in the investigated copolymers are necessary.
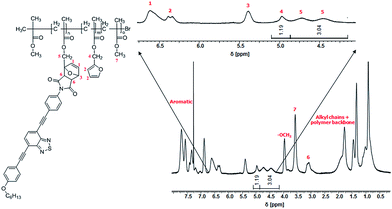 |
| | Fig. 1 Quantification of free furan moieties in the polymer back bone via1H NMR spectrum (CDCl3) of acceptor graft copolymer P2. | |
Copolymer synthesis via direct polymerization
However, for the subsequent exchange studies between donor and acceptor functionalized graft copolymers, it would be desirable that no free furan moieties were present in the copolymers. Thus, MMA was copolymerized with acceptor oligomer methacrylate 20 by a standard free radical polymerization technique in a ratio of MMA:20 of 5.6:1 (Scheme 3). The temperature during the polymerization process was kept low due to the reversibility of the Diels–Alder functionality at around 70 °C. 2,2′-Azobis(4-methoxy-2,4-dimethyl valeronitrile) (V70) was utilized as azo initiator. The same procedure has been applied to the donor oligomer methacrylate 21. Further details are summarized in ESI Scheme 4.† The obtained molar mass and Đ values of acceptor side chain copolymer P4 and donor side chain copolymer P5 are summarized in Table 1. The Mn values of copolymers P4 and P5 are smaller than those of copolymers P2 and P3, respectively.
 |
| | Scheme 3 Schematic representation of the polymer synthesis of acceptor containing graft copolymer P4. Reagents and conditions: (i) dimethylformamide/40 °C. | |
In contrast to the copolymers obtained via the “grafting to method”, no free furan moieties were detectable in the 1H NMR spectrum of copolymer P4 and a retro Diels–Alder reaction of the oligomer during polymerization can thus be excluded (Fig. 2). Furthermore, the resulting monomer ratio of MMA:20 of 4.3:1 was calculated via1H NMR spectrum. Copolymer P5 consists of a similar monomer ratio of MMA:21 of 4.3:1, which was calculated from 1H NMR spectrum in ESI Fig. 3.†
 |
| | Fig. 2 Quantification of polymerized monomer ratio and free furan groups via1H NMR spectrum (CDCl3) of acceptor containing graft copolymer P4. | |
Optical properties
The oligomers 20 and 21 as well as their corresponding copolymers P4 and P5 have been characterized by means of steady-state UV-vis absorption and emission spectroscopy (Fig. 3). Comparing the normalized absorption spectra of the copolymers and their respective monomer, they are near identical and no shift of the respective long-wavelength absorption maximum was observed (Table 2). This indicates lack of interactions of the electronic ground-states of the chromophores, e.g., π–π-stacking. A slightly increased absorption below 300 nm could be seen for both copolymers and is attributed to contributions of the copolymer backbone. Similarly, no pronounced differences in the emission spectra of the copolymers and their respective monomers could be observed. The emission maxima remain virtually unchanged, while a small spectral broadening in the low-energy region of their emission spectra could be observed for both copolymers.
 |
| | Fig. 3 UV-vis absorption and fluorescence spectra (λex = 369 nm) of acceptor monomer 20/copolymer P4 and donor monomer 21/copolymer P5 in CHCl3. Absorption coefficients for the copolymers refer to moles of side-chain chromophores, assuming a MMA:chromophore ratio of 4.3:1. Absorption spectra of the copolymers normalized to the lowest energy absorption maximum of their respective monomer are additionally shown as dashed lines. | |
Table 2 Steady state data on absorption and emission of the copolymers and oligomers under consideration. The molar absorption coefficient ελ,max, referring to moles of chromophores, at the lowest energy peak position λabs,max as well as the peak wavelength of fluorescence λfl,max together with the calculated Stokes shift Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) and the fluorescence quantum yield Φ
and the fluorescence quantum yield Φ
| Compound |
λ
abs,max (nm) |
ε
λ,max (M−1 cm−1) |
λ
fl,max (nm) |
Δ (cm−1) |
Φ
|
|
20
|
428 |
28200 |
538 |
4777 |
0.90 |
|
P4
|
428 |
23800 |
541 |
4880 |
0.56 |
|
21
|
372 |
40800 |
409 |
2360 |
0.80 |
|
P5
|
372 |
43800 |
410 |
2419 |
0.64 |
Comparison of the absorption spectra of acceptor monomer 20 with copolymer P4 shows that the molar absorption coefficient (referring to moles of monomer incorporated in the copolymer) of the copolymer, which has been calculated assuming a ratio MMA:20 of 4.3:1 (see above), is lower than of its monomer counterpart. In similar systems, no pronounced change in oscillator strength of the chromophore could be observed, when it was attached to a polymer backbone.29 As such, assuming that this is also the case in the systems under consideration, one can deduce a MMA:20 ratio of 6.4:1 for copolymer P4. Similarly, a MMA:21 ratio of 3.3:1 for copolymer P5 was calculated from the absorption spectra.
Exchange studies between oligomers and graft copolymers
In order to investigate the reversibility of the introduced Diels–Alder functionality and the resulting exchange of the conjugated oligomers, first, the exchange between free oligomeric side chains and graft copolymers was studied by means of 1H NMR spectroscopy and SEC measurements (diode array detector). For this purpose, the acceptor oligomer 12 was dissolved in chlorobenzene and heated up to 120 °C for about four hours, resulting in evaporation of furan moieties due to a retro Diels–Alder reaction occurred. Subsequently, a mixture of prepared 6.25 wt% maleimide functionalized acceptor oligomer 12 and 93.75 wt% donor graft copolymer P5 dissolved in chlorobenzene was drop casted resulting in well-defined polymer films, which were heated up to 67 °C for four days. The temperature of 67 °C was determined as the ideal condition for equilibrium between Diels–Alder and retro Diels–Alder reaction in the prepared systems. The resulting exchanged oligomer/copolymer mixture was qualified via SEC measurement with diode array detector (Fig. 4). Fig. 4a show the SEC measurement before the exchange reaction. At an elution volume of 18.6 mL, only donor moieties in the copolymer fraction are visible. In contrast, at an elution volume of approximately 22.1 mL (oligomer fraction), only acceptor moieties are visible. In contrast, Fig. 4b depicts the SEC measurement after heating up to 67 °C for four days. In the oligomer as well as in the copolymer fraction, spectral features of donor and acceptor moieties are present. This is indicative for an exchange of the side chains between acceptor oligomer 12 and donor containing graft copolymer P5.
 |
| | Fig. 4 SEC measurements with diode array detector: starting points of (a) donor graft copolymer P5/acceptor oligomer 12 and after exchange in polymer film (b) mixture of acceptor–donor graft copolymer and donor/acceptor oligomer. | |
The same procedure was carried out for the exchange of donor oligomer 19 and acceptor containing graft copolymer P4. The results are depicted in ESI Fig. 4.† Similar to Fig. 4b, spectral features of both donor and acceptor moieties in copolymer and oligomer fraction after exchange procedure could be observed. Furthermore, the exchange was quantified via calculation from 1H NMR spectra of the resulting copolymers. After heating up to 67 °C for about four days, the polymer film was dissolved in chloroform and purified via Biobeads® (S-X1) in order to separate the non-converted oligomer fraction. The 1H NMR spectra of the acceptor containing graft copolymer P4 before and after exchange are depicted in Fig. 5. Approximately 0% of free furan moieties exist in the donor–acceptor graft copolymer after exchange due to the presence of a small access of free side chain oligomers during the exchange process. It was calculated, that approximately 17% of the donor moieties in the donor containing graft copolymer P5 were exchanged with acceptor chromophores. Similarly, the exchange quantity between donor containing graft copolymer P5 and acceptor oligomer 12 amounted to about 16% as calculated from the 1H NMR measurement. Further details are summarized in ESI Fig. 5.†
 |
| | Fig. 5 Quantification of donor and acceptor dye ratio and free furan groups via1H NMR spectrum (CDCl3): (a) before and (b) after exchange between acceptor containing graft copolymer P4 and donor oligomer 19. | |
Exchange studies between graft copolymers
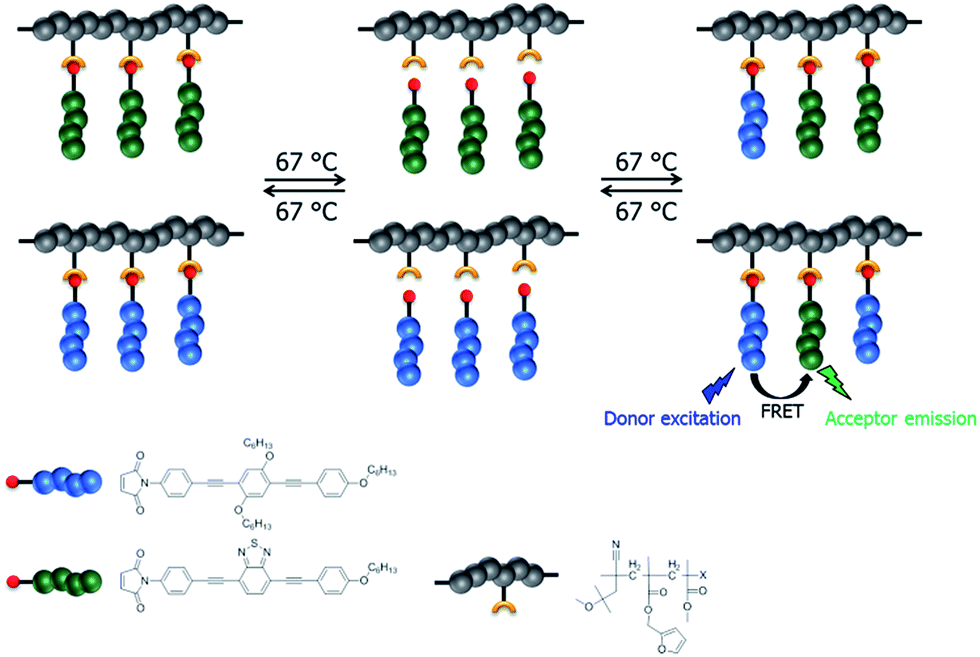
Finally, exchange of the side chain oligomers between the prepared acceptor containing graft copolymer and the donor containing graft copolymer has been investigated. A schematic overview of the general procedure of the exchange studies and the utilized materials is depicted in Scheme 4.
 |
| | Scheme 4 Schematic representation of the utilized materials and the general procedure of the exchange study between prepared graft copolymers. | |
The donor graft copolymer P5 and the acceptor graft copolymer P4 were dissolved in chlorobenzene in a mass ratio of 1:1 and, subsequently, drop casted to obtain a polymer film. After heating up to 67 °C for about four days, the polymer film was dissolved in chloroform and purified via Biobeads® (S-X1) in order to separate the split off oligomer fraction. The quality of exchange and the quantity of free furan moieties after exchange was determined via calculation by NMR techniques of the prepared mixed donor–acceptor graft copolymer. Thereby, DOSY NMR represents an interesting technique for proof of exchange. The proof of clicking two polymer chains by Diels–Alder30 or “grafting to” by Diels–Alder31 was already investigated by DOSY NMR, but to the best of our knowledge not for exchange of graft copolymers. However, DOSY NMR measurements indicate an exchange of the chromophores between the two graft copolymers (Fig. 6). P4 and P5 exhibit different diffusion coefficients (Fig. 6a and b). After exchange, a statistical donor–acceptor copolymer species with a separated new diffusion coefficient was obtained (Fig. 6c). The resulting 1H NMR spectrum includes signals of both graft copolymers; in particular, the separated signal of P4 at 7.75 ppm and the signal of P5 at 7.48 ppm (ESI Fig. 6†). This leads to the conclusion that a statistical donor–acceptor graft copolymer was yielded during the heating process. Based on this knowledge, the exchange was quantified via1H NMR measurement (Fig. 7). The calculation indicates that 14% of furan groups are not converted and approximately 43% are donor oligomer and 43% are acceptor oligomer connected.
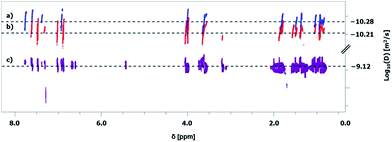 |
| | Fig. 6 DOSY-NMR spectra of (a) P4, (b) P5 and (c) after exchange of P4 and P5 (CDCl3). | |
 |
| | Fig. 7 Quantification of donor and acceptor dye ratio and free furan groups via1H NMR spectrum (CDCl3) after exchange between acceptor containing graft copolymer P4 and donor containing graft copolymer P5. | |
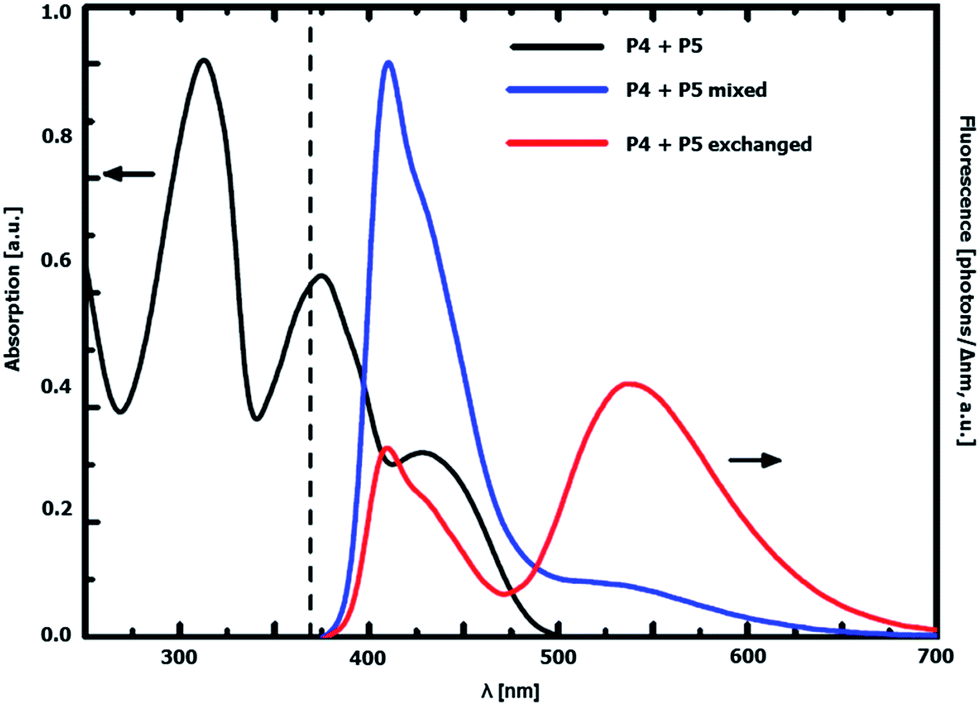
The interaction of the rigid acceptor and donor dyes in the exchanged graft copolymer was further investigated via UV-vis measurements. The absorption and emission spectra of a mixture of graft copolymer P4 and P5 before exchange (50:50 wt%) and the obtained donor–acceptor graft copolymer after exchange are depicted in Fig. 8. The absorption spectra of both the mixture of the copolymers as well as the sample after exchange do not differ from each other, which indicates a very similar composition of both and a lack of ground-state interaction between donor and acceptor side chains in the copolymer after exchange. However, a change in the emissive behavior could be observed. The emission of the copolymer mixture upon 369 nm excitation, i.e. exciting near the absorption maximum of the donor, mostly consists of donor contributions. Acceptor emission results from direct absorption of graft copolymer P5 at the excitation wavelength.
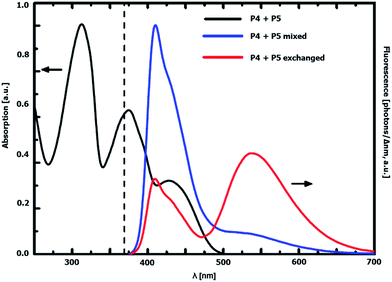 |
| | Fig. 8 UV-vis absorption (black) and emission spectra (λexc = 369 nm, dashed line) of a mixture of P4 and P5 before exchange (blue) and the statistical donor–acceptor graft copolymer after exchange (red) in CHCl3. | |
On the other hand, the exchange product contains substantial acceptor emission under the same experimental conditions. This can be explained by an inter side chain energy transfer from the donor to the acceptor side chains taking place, as there is a considerable overlap between donor emission and acceptor absorption (Fig. 3). From the steady-state spectra, a Förster radius R0 = 43 Å could be derived, which is still large enough for fluorescence resonance energy transfer (FRET) to occur in the system investigated: from the UV-vis data of graft copolymers P4 and P5 and the NMR data after exchange, it is possible to estimate an average donor–acceptor distance. According to these, the average distance between donor and acceptor amounts to 37 Å in the exchanged copolymer (see ESI† for a detailed explanation).
By means of time-resolved emission spectroscopy, the efficiency of energy transfer and, consequently, the average distance of donor and acceptor chromophores could be calculated. In the case of donor monomer 21 and donor containing graft copolymer P5, no difference in emission lifetimes was observed; τ21 = τP5 = 1.05 ns (Fig. 9a). However, in the exchanged copolymer, donor emission is quenched due to FRET occuring. In contrast to donor monomer 21 and donor containing graft copolymer P5, emission in the exchanged copolymer does not mono-exponentially decay (Fig. 9a, inset). This indicates that there is not a single donor–acceptor distance, but a distribution of distances.32 This finding is in line with the assumption that the exchange yields a statistical copolymer. By fitting the data using a probability weighted average (assuming a Gaussian distribution), a mean donor–acceptor distance of R = 51 Å (FWHM = 20 Å) was calculated. On the other hand, acceptor monomer 20 (τ20 = 4.68 ns), acceptor containing graft copolymer P4 (τP4 = 4.15 ns) and the acceptor in the exchanged copolymer (τA,exc = 3.86 ns) exhibit very similar emission lifetimes (Fig. 9b). The small differences might originate from minor inter side chain interactions, which were not detectable from the steady-state data.
 |
| | Fig. 9 Time resolved emission data recorded after 380 nm excitation for (a) donor emission between 400 and 450 nm (inset: zoom-in to region of interest at small times) and (b) acceptor emission between 520 and 570 nm in CHCl3. All decay curves have been normalized to their respective intensity maximum. | |
Conclusions
In summary, the synthesis and characterization of graft copolymers with oligomeric side chains was realized using “grafting to” as well as free radical polymerization technique. Moreover, a Diels–Alder functionality was introduced between the oligomeric side chains and the polymer backbone, which exhibits reversibility of the side chains at moderate temperatures (67 °C). In this contribution, the exchange of the conjugated oligomers was investigated between oligomers and graft copolymers as well as between two different graft copolymers in solid state by thermal treatment of the polymer films. The exchange was proven by DOSY NMR spectroscopy, size exclusion chromatography as well as UV-vis measurements and quantified by 1H NMR spectroscopy. The resulting donor–acceptor graft copolymer exhibits the same ground state properties as the mixture of donor containing graft copolymer and acceptor containing graft copolymer before exchange, whereby the emission properties of the donor–acceptor graft copolymer were changed significantly due to intra polymer FRET.33 In contrast to known graft copolymers with implemented π-conjugated side chains, this novel approach opens the possibility to initiate an energy transfer between donor and acceptor side chains connected with changed emission properties by thermal triggering. Therefore, the prepared system can be potentially applied for optical tuning of π-conjugated polymer films after processing as well as applications as thermally triggered sensor systems.
Acknowledgements
The authors would like to thank the Deutsche Forschungsgemeinschaft (DFG) for financial support within the framework of the priority program SPP1568 (Design and Generic Principles of Self-healing materials; HA6306/3-1, DI1517/9-1) as well as Dr Peter Bellstedt for DOSY NMR measurements.
Notes and references
- C. Feng, Y. J. Li, D. Yang, J. H. Hu, X. H. Zhang and X. Y. Huang, Chem. Soc. Rev., 2011, 40, 1282–1295 RSC.
- C. Weber, C. R. Becer, W. Guenther, R. Hoogenboom and U. S. Schubert, Macromolecules, 2010, 43, 160–167 CrossRef CAS.
- T. Pakula, Y. Zhang, K. Matyjaszewski, H. I. Lee, H. Boerner, S. H. Qin and G. C. Berry, Polymer, 2006, 47, 7198–7206 CrossRef CAS.
- Z. Ying, N. Costantini, M. Mierzwa, T. Pakula, D. Neugebauer and K. Matyjaszewski, Polymer, 2004, 45, 6333–6339 CrossRef.
- J. Rzayev, Macromolecules, 2009, 42, 2135–2141 CrossRef CAS.
- J. Plank, Z. M. Dai and N. Zouaoui, J. Phys. Chem. Solids, 2008, 69, 1048–1051 CrossRef CAS.
- P. Banerjee, D. J. Irvine, A. M. Mayes and L. G. Griffith, J. Biomed. Mater. Res., 2000, 50, 331–339 CrossRef CAS PubMed.
- J. H. Hyun, H. W. Ma, Z. P. Zhang, T. P. Beebe and A. Chilkoti, Adv. Mater., 2003, 15, 576–579 CrossRef CAS.
- G. M. Pavlov, A. M. Breul, M. D. Hager and U. S. Schubert, Macromol. Chem. Phys., 2012, 213, 904–916 CrossRef CAS.
- Y. Shirota, J. Mater. Chem., 2000, 10, 1–25 RSC.
- R. Schroot, U. S. Schubert and M. Jäger, Macromolecules, 2015, 48, 1963–1971 CrossRef CAS.
- R. Schroot, C. Friebe, E. Altuntas, S. Crotty, M. Jäger and U. S. Schubert, Macromolecules, 2013, 46, 2039–2048 CrossRef CAS.
- J. Kübel, R. Schroot, M. Wächtler, U. S. Schubert, B. Dietzek and M. Jäger, J. Phys. Chem. C, 2015, 119, 4742–4751 Search PubMed.
- C. C. Zhao, Y. Zhang, S. L. Pan, L. Rothberg and M. K. Ng, Macromolecules, 2007, 40, 1816–1823 CrossRef CAS.
- Y. Shirota, T. Nogami, T. Kakuta and H. Saito, Synth. Met., 1991, 41, 1169–1172 CrossRef CAS.
- I. R. Jeon, N. Noma and Y. Shirota, Bull. Chem. Soc. Jpn., 1992, 65, 1062–1066 CrossRef CAS.
- A. M. Breul, J. Schafer, G. M. Pavlov, A. Teichler, S. Hoppener, C. Weber, J. Nowotny, L. Blankenburg, J. Popp, M. D. Hager, B. Dietzek and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3192–3205 CrossRef CAS.
- M. Obi, S. Morino and K. Ichimura, Chem. Mater., 1999, 11, 1293–1301 CrossRef CAS.
- A. Wild, A. Teichler, C. L. Ho, X. Z. Wang, H. M. Zhan, F. Schlutter, A. Winter, M. D. Hager, W. Y. Wong and U. S. Schubert, J. Mater. Chem. C, 2013, 1, 1812–1822 RSC.
- R. Abbel, C. Grenier, M. J. Pouderoijen, J. W. Stouwdam, P. E. L. G. Leclere, R. P. Sijbesma, E. W. Meijer and A. P. H. J. Schenning, J. Am. Chem. Soc., 2009, 131, 833–843 CrossRef CAS PubMed.
- X. X. Chen, M. A. Dam, K. Ono, A. Mal, H. B. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- X. X. Chen, F. Wudl, A. K. Mal, H. B. Shen and S. R. Nutt, Macromolecules, 2003, 36, 1802–1807 CrossRef CAS.
- Y. Zhang, A. A. Broekhuis and F. Picchioni, Macromolecules, 2009, 42, 1906–1912 CrossRef CAS.
- J. Kötteritzsch, S. Stumpf, S. Hoeppener, J. Vitz, M. D. Hager and U. S. Schubert, Macromol. Chem. Phys., 2013, 214, 1636–1649 CrossRef.
- R. Pizzoferrato, M. Berliocchi, A. Di Carlo, P. Lugli, M. Venanzi, A. Micozzi, A. Ricci and C. Lo Sterzo, Macromolecules, 2003, 36, 2215–2223 CrossRef CAS.
- B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci and A. L. Demirel, Macromolecules, 2006, 39, 5330–5336 CrossRef CAS.
- L. Xiao, Y. Chen and K. Zhang, Macromolecules, 2016, 49, 4452–4461 CrossRef CAS.
- A. Bousquet, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1773–1781 CrossRef CAS.
- J. Schäfer, A. Breul, E. Birckner, M. D. Hager, U. S. Schubert, J. Popp and B. Dietzek, ChemPhysChem, 2013, 14, 170–178 CrossRef PubMed.
- C. F. Hansell, P. Espeel, M. M. Stamenovic, I. A. Barker, A. P. Dove, F. E. Du Prez and R. K. O'Reilly, J. Am. Chem. Soc., 2011, 133, 13828–13831 CrossRef CAS PubMed.
- L. Billiet, O. Gok, A. P. Dove, A. Sanyal, L.-T. T. Nguyen and F. E. Du Prez, Macromolecules, 2011, 44, 7874–7878 CrossRef CAS.
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4588 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21536j |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence