
Preparation and application of surface activated Si-MCM-41 and SBA-16 as reusable supports for reduction of cyclic ketones with preferential stereoselectivity†
Abstract
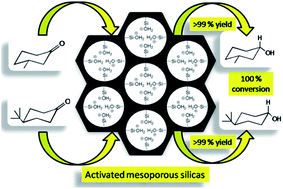
An efficient and benign procedure for the reduction of a few cyclic ketones adsorbed on the activated surface of calcined Si-MCM-41 and calcined SBA-16 using NaBH4 as the reducing agent with ethanol as the medium resulted in the formation of two epimeric alcohols. After synthesis, these calcined materials were treated with concentrated HCl to activate surface silanol groups. Various instrumental techniques like FTIR, XRD, N2 sorption isotherms, FESEM, TEM and XPS were carried out to examine the pre and post activated surface of the used supports. Five cyclic ketones were reduced. Reduction of 4-tert-butylcyclohexanone yielded only trans-4-tert-butylcyclohexanol. Moreover, exclusive formation of cis-3-methylcyclohexanol (equatorial –OH) was also observed. This work offers several advantages such as a simple operational procedure, short reaction time, and high yield of the product, along with maintaining the materials' diversity. This is due to the presence of the activated silanol groups of these materials, which cause nucleophilic activation of the carbonyl group of ketones leading to faster reaction rates. Beside this, these supports can be regenerated well from the reaction mixture using a calcination treatment followed by concentrated HCl, and reused several times without causing any serious malformation in the activated surfaces. Finally, this work opens up a new direction of research for the fabrication of solid reusable supports in the reduction of cyclic ketones.
 Please wait while we load your content...
Please wait while we load your content...

