
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Allylation of a lignin model phenol: a highly selective reaction under benign conditions towards a new thermoset resin platform†
M. Jawertha,
M. Lawokoa,
S. Lundmarkab,
C. Perez-Berumenac and
M. Johansson*a
aWallenberg Wood Science Center, WWSC, Department of Fibre and Polymer Technology, KTH Royal Institute of Technology, 100 44 Stockholm, Sweden. E-mail: matskg@kth.se
bPerstorp AB, 284 80 Perstorp, Sweden
cDepartment of Organic Chemistry, Universidad Autonoma de Coahuila, Faculty of Chemical Sciences, Blvd. V. Carranza y J. Cardenas, 25280 Saltillo, Coah, Mexico
First published on 4th October 2016
Abstract
The lack of aromatic material constituents derived from renewable resources poses a problem to meet the future demands of a more sustainable society. Lignin is the most abundant source of aromatic structures found in nature and is a highly interesting source for material applications. Development of controlled chemical modification routes of lignin structures are crucial in order to further develop this area. In this study allyl chloride is used to selectively modify a lignin phenol in the presence of other lignin functionalities, i.e. aliphatic hydroxyls and conjugated alkenes, under mild reaction conditions in quantitative yields. For this, coniferyl alcohol was used as a model compound in the present study. The modification was carried out in ethanol as the synthesis media. Studies on the effect of reaction time and temperature revealed optimum conditions allowing for a quantitative yield without any detectable levels of byproducts as studied with NMR, FT-IR and FT-Raman. The thermal stability of the formed product was determined to be up to at least 160 °C through DSC measurements. In addition, as a proof of concept, the use of the allylated monomer to form crosslinked films using free radical thiol–ene polymerization was demonstrated.
Introduction
The development efforts toward new materials based on renewable resources continuously increase to address the future demands of a more sustainable society.1 A wide range of resources are exploited as replacements for fossil based material constituents in order to achieve this goal.2 One of the biggest challenges is to find alternatives to aromatic structures currently used and only a few examples have been introduced as replacements in conventional polymer systems.3–5 Furan dicarboxylic acid, made from sugars, is for example now produced as an alternative to terephthalic acid.6 The most abundant source for natural aromatic structures is however lignins found in lignocellulosic biomass.3,7,8 Lignin is a macromolecular constituent in plants that has gained an increased interest in the past 30 years due to its low cost, its abundance and the drive to find new material applications derived from renewable resources.7–10 It is the largest source of naturally occurring phenolic compounds and offers a potential alternative for non-renewable aromatic structures.7 Many studies in this field have, since the 60s, been performed to use lignin as a polymeric material in thermoplastic and thermoset resin applications.8,10The main production source that exist for extraction of lignins are the pulp and paper processes, which globally produce about 70 million tonnes of lignin every year.7,11 However only 2% are utilized for commercialization,8 the rest is combusted in a chemical recovery boiler producing energy due to its high heating value of about 27 MJ kg−1.12 The growing consensus however, is that part of the lignin can be extracted for production of bio-based products and create value addition for existing industries.7,8 A few techniques, such as the lignoboost technology, which was recently commercialized, are emerging to allow lignin with relatively high purity to be obtained in powder form.11–13 However, the polydispersity of lignin with regards to molar mass distribution is high. Lignoboost kraft lignin for example will have a polydispersity, Ð, of more than six.13
Difficulties in utilizing lignin as a resource is not only due to the molecular weight distribution but also on the abundance of different functional groups dependant on the process applied for the extraction.7,11,13 The challenge in using lignin and lignin derivatives as material building blocks thus includes fractionation, characterization, and modification of complex systems to obtain suitable target constituents for material applications.7,8,14,15 Fractionation using solvent sequences has therefore been considered as one route to obtain more homogeneous fractions to address these problems.11,13
Variations are also largely depending on the specie used as the lignin source. Lignins have a composition comprised of phenolic monomers referred to as monolignols.7 There are mainly three different monolignols distinguished by their degrees of substitution by methoxy groups in the meta positions of the aromatic ring.7,9 These are called p-coumaryl alcohol, coniferyl alcohol and sinapyl alcohol8,9,15,16 respectively, Fig. 1.
 | ||
| Fig. 1 The three different monolignols, from left to right: p-coumaryl alcohol, coniferyl alcohol and sinapyl alcohol labelled according to established literature. | ||
Depending on plant species, the lignin contains different proportions of the three monophenolic compounds. Softwood lignins consist majorly of coniferyl alcohol, hardwood lignins of a mixture between sinapyl alcohol and coniferyl alcohol and in grass the lignin is comprised of all three types of monolignols.7,8 The exact structure and polymerization degree of lignin in plants however remain unknown although extensively studied.14,17
Lignin molecules are equipped with different functionalities, most of them being hydroxyl groups of phenolic or aliphatic nature. Technical lignin can however carry other kind of functionalities such as alkene, carboxyl, or carbonyl groups.7,8,11 Due to this diversity and complexity in structure, lignin provides a challenge in terms of handling and application.7
Modification of lignin can be performed on the extracted macromolecular lignin, oligomeric lignin or mono phenols to match the various application demands.7,15,18 One of few lignin derived pure compounds produced technically in large volumes is vanillin, which have been studied for material applications i.e. by Fache et al.,3 Meylemans et al.4 and Harvey et al.5 Depending on end use, the modification of the phenolic hydroxyl groups present in lignin structures could be desirable since these are weakly acidic and relatively easily oxidized, consequently causing deterioration and discoloration of the final material if still present.19,20 An efficient and selective modification of phenols in the presence of other functional groups such as aliphatic alcohols and alkenes is thus highly desirable and would allow for use of more complex lignin structures as material precursors.
One specific reaction that has been performed to modify lignin and lignin model compounds is allylation to form allyl ethers.10,21–23 Allyl compounds, first identified, named and described in 1844 24 have ever since been used as base structures for various polymer systems either by using it directly in polymer forming reactions or as a precursor for other reactive moieties.25,26 Allyl mercaptan was for example the monomer used when first describing the thiol–ene reaction as a polymer forming reaction.27 The thiol–ene reaction has been drawing attention in recent years due to its “click” characteristics under certain reaction conditions, different possible reaction mechanisms, and structural versatility.28–30 The allyl functional group also open up for a large variety of other different synthetic pathways as described in literature in numerous papers.21,23,31,32,33,34 This can, if applied to lignin structures, be considered as a material platform as shown in Scheme 1.
 | ||
| Scheme 1 Different proposed reaction pathways for utilization of allylated lignin structures. | ||
One other interesting route to consider in thermoset material applications is the epoxidation of the double bond to form glycidyl ethers.23 Around 70% of the market of thermosetting resins, excluding polyurethanes, is made up of epoxy resins which is used in many different industrial applications with high material demands.15 Epoxy resins are versatile and can be found in coatings, adhesives and high performance composites. Because of the convenience of epoxy resins there are high interests in finding resin systems, partially or fully, derived from renewable resources. Combining the epoxy functionality and the aromaticity of lignin is therefore highly interesting for future efforts.15
Allylation of lignin using different allyl compounds have been described in the literature.10,21–23 Allyl halides in the form of allyl bromide and allyl chloride has been utilized by Zoia et al.21 and Dournel et al.10 respectively to form allyl ethers on lignin and lignin model compounds. Dournel et al.10 examined the allylation reaction on many low molecular weight lignin model compounds in various yields ranging between 35 and 100%. However, their model compounds all contain either a phenolic hydroxyl group or an aliphatic one, never the two types within the same compound. Zoia et al.21 reported an overall conversion yield of 80–90% when performing the allylation reaction on guaiacylic lignin in acetone and an excess of allyl bromide in relation to the total amount of hydroxyl groups. Over et al.22 have on the other hand examined the possibilities of using diallyl carbonate as a more environmentally friendly reagent. They found that 99% of the phenolic hydroxyl groups were allylated while 71% of the aliphatic were converted resulting in an overall yield of 82%. In all three mentioned papers it is thus hinted that the phenolic hydroxyl group is more prone to be modified than those of aliphatic nature, however a fully selective reaction is yet to be shown.
The present study describes a route to selectively modify phenol groups in a lignin model compound to a very high conversion under mild reaction conditions using an environmentally friendly solvent. In addition, as a proof of concept, crosslinked molecular networks were prepared using thiol–ene chemistry and analysed.
Experimental
Materials
All chemicals were of analytical grade and used as received.Allyl chloride (98%), coniferyl aldehyde (98%), sodium borohydride (NaBH4, 98%), sodium carbonate (Na2CO3, 99.5%), sodium hydroxide (NaOH, ≥98%) and trimethylolpropane tris(3-mercaptopropionate) (TMP, 95%) was purchased from Sigma Aldrich. Ethanol (96%), ethyl acetate (EtOAc), dichloromethane (DCM), heptane and chloroform were obtained through VWR chemicals. Magnesium sulphate (MgSO4, 99%) was bought from Acros Organics and sodium chloride (NaCl) from Merck. Diethyl ether was achieved through Fisher Scientific. Irgacure 651 was obtained from BASF chemicals.
Methods
Reduction of coniferyl aldehyde to coniferyl alcohol (CA)
Coniferyl alcohol (CA) was prepared from coniferyl aldehyde based on procedures previously reported by Ralph et al.35,36NaBH4 (1.28 g, 33.7 mmol) was dispersed in 180 ml of EtOAc in a 500 ml round bottom flask under agitation. Coniferyl aldehyde (3.00 g, 16.8 mmol) was added to the reaction vessel and the mixture was left to stir for 17.5 hours in room temperature. 200 ml of deionized water was carefully added to quench the reaction and the mixture was agitated for 30 additional minutes. The organic phase was extracted and washed with brine before being dried using MgSO4 and filtered. The EtOAc was removed in a rotary evaporator at 40 °C. An oily bright yellow product was obtained and re-dissolved in warm DCM before being stored in a freezer to recrystallize for roughly 1.5 hours. The product was filtered and washed with heptane before being dried in vacuum oven at 50 °C overnight. The leftover mixture from the crystallization step was dried using a rotary evaporator at 40 °C and the procedure was repeated and left over night to yield more product. The result was white crystals with an overall yield of ≈80%. 1H-NMR (400 MHz, DMSO-d6) δ 8.98 (s, 1H), 6.99 (d, J = 1.9 Hz, 1H), 6.79 (dd, J = 8.2, 2.0 Hz, 1H), 6.70 (d, J = 8.1 Hz, 1H), 6.41 (dt, J = 15.8, 1.6 Hz, 1H), 6.17 (d, J = 15.9 Hz, 0H), 4.75 (t, J = 5.5 Hz, 1H), 4.07 (td, J = 5.5, 1.7 Hz, 2H), 3.78 (s, 3H). 13C-NMR (101 MHz, DMSO-d6) δ 147.72, 146.17, 129.00, 128.52, 127.50, 119.44, 115.48, 109.70, 61.75, 55.56.
Synthesis of coniferyl alcohol allyl ether (CAAE)
NaOH (0.32 g, 7.77 mmol) was dissolved in 30 ml of ethanol under agitation while heated to 55 °C. A condenser was applied to prevent evaporation of solvent and allyl chloride. When the temperature had reached 55 °C and the NaOH was dissolved, CA (1 g, 5.55 mmol) was added to the mixture and dissolved. Allyl chloride (0.63 ml, 7.77 mmol) was carefully provided to the solution using a syringe through a septum and the reaction was left to stir for 17.5 hours. The reaction was terminated by stopping the flow in the condenser and open up the system for 30 min to let excess allyl chloride evaporate. Ethanol was rotaevaporated at 40 °C. The yellow viscous product was re-dissolved in diethyl ether and extracted towards a 10 wt% Na2CO3 water solution. The organic phase was dried using MgSO4 and filtered. Diethyl ether was removed using a rotary evaporator at 30 °C. The product was dried in vacuum oven at 50 °C over night and the yield was determined to 80%. 1H-NMR (400 MHz, DMSO-d6) δ 7.05 (t, J = 1.1 Hz, 0H), 6.89 (d, J = 1.4 Hz, 1H), 6.53–6.39 (m, 0H), 6.34–6.19 (m, 0H), 6.11–5.92 (m, 0H), 5.38 (dd, J = 17.3, 1.8 Hz, 0H), 5.24 (dd, J = 10.5, 1.7 Hz, 0H), 4.81 (t, J = 5.5 Hz, 0H), 4.53 (dt, J = 5.4, 1.5 Hz, 1H), 4.09 (td, J = 5.4, 1.6 Hz, 1H), 3.78 (s, 1H). 13C-NMR (101 MHz, DMSO-d6) δ 149.16, 147.10, 133.87, 130.21, 128.68, 128.50, 119.03, 117.45, 113.46, 109.44, 68.95, 61.63, 55.48.Conversion as a function of time
The experimental procedure was the same as for the first CAAE synthesis but in 2.77 mmol (≈0.5 g) scale.0.7 ml of the solution was removed, dried using vacuum and analysed by 1H-NMR. This was repeated during the course of the reaction to be able to determine the conversion for each point in time.
Effect of reaction temperature
The experimental procedure was the same as for the first CAAE synthesis but in 1.66 mmol (≈0.3 g) scale with respect to everything but the solvent, 15 ml was used for practical reasons.The reaction proceeded for 6 hours before 0.7 ml was removed, dried using vacuum and used for 1H-NMR analysis to determine the conversion. This was repeated when 24 hours had past. The procedure was performed for the temperatures 25, 35, 45, 55, 65, 75 and 85 °C to evaluate the effect of temperature on the reaction rate. The NMR-tubes were cooled with dry ice in acetone before each sample was extracted to quickly reduce the reaction speed. Vacuum was applied to remove the residual allyl chloride and ethanol and the dried mixture were redissolved in DMSO before the NMR analysis was performed.
Thermoset crosslinking through thiol–ene coupling
CAAE, TMP and Irgacure 651 (≈3 wt%) was added to a glass vial and mixed using ≈100 mg chloroform. The resin was spread out on a glass substrate and chloroform left to evaporate for 10–15 minutes before the mixture was covered using a glass cover. The resins were placed under UV-light for 30 minutes to cure. The resulting films was removed from the glass and retrieved for analysis. Three films were prepared with different thiol/ene molar ratios with regards to reacting groups (+10 thiol, equimolar ratio, and −10% thiol). The cured films were analysed by FT-IR, FT-Raman and DSC.Result and discussion
The purpose of this study was to establish a synthesis route to selectively modify the phenol group in a lignin model compound in the presence of primary alcohols and conjugated alkenes, reaction scheme in Scheme 2. This will provide a tool for modification of more complex lignin derivatives with an overall aim to create new thermoset resin platforms. Coniferyl alcohol was selected as a model compound since it is the main monolignol in softwood7 and contain 3 important lignin functionalities; a conjugated alkene, a phenol and an aliphatic hydroxyl. In addition, it could be easily synthesized from the cheaply available aldehyde. Allyl chloride was chosen for the modification since it has suitable reactivity as will be discussed later. In addition, it is a well-established chemical used for allylation of aliphatic structures in an industrial scale.37 Ethanol was used as a solvent for several reasons: all reagents are soluble; it is environmentally friendly and easy to handle. Through solvent sequence fractionation it has also been shown that more than 40% of lignoboost kraft lignin is soluble in ethanol and render a fraction with a Ð of about 2, which is promising for both future academic work as well as for industrial purposes.7,13 Furthermore, the reaction was performed under ambient atmosphere to verify that atmospheric oxygen has no effect on the reaction. This would also mimic the atmosphere in true technical processes. | ||
| Scheme 2 A selective modification of coniferyl alcohol (CA) using allyl chloride to form coniferyl alcohol allyl ether (CAAE). | ||
Reaction conditions, including time and temperature dependences, were studied for the synthesis. The synthesized monomers thermal properties were also examined and analysed for both characterization purposes and to determine how the product can be handled for further functionalization and utilization. The in house synthesis of the model compound, CA, as described above, granted purity of the reagent36 since coniferyl alcohol has an intrinsic instability in the structure making it prone to oxidation to coniferyl aldehyde when stored, also observed in our laboratory.
As a proof of concept, crosslinked thermoset films were prepared through free radical thiol–ene crosslinking using TMP as a trifunctional thiol functional crosslinker, Scheme 3. The modified model compound would in this case act as a bi-functional monomer where both the allyl ether as well as the α–β double bond allow crosslinked thermosets to be made.
 | ||
| Scheme 3 Thiol–ene crosslinking using TMP and CAAE. | ||
Synthesis of CAAE
Characterization and validation for the synthesis were carried out by 1H-, 13C & 2D HSQC NMR, FT-IR, FT-Raman and GC-MS.1H- & 13C-NMR
1H-NMR reveals the difference in structure between CA and the formed product, Fig. 2. One can first notice that the peak corresponding to the phenol proton at ∂ = 8.98 ppm has disappeared in the product while the primary alcohol triplet peak at ∂ = 4.75 ppm is still present, only shifted to ∂ = 4.81 ppm. Three additional peaks of interest have appeared, one double triplet at ∂ = 4.53 ppm, two double quadruplets at ∂ = 5.24 ppm and ∂ = 5.38 ppm and one characteristic multiplet at ∂ = 6.03 ppm. These are peaks assigned to the newly formed allyl ether. It is also worth noticing that the double bond between the α (∂ = 6.41 ppm) and β (∂ = 6.18) carbons are preserved and not affected by the reaction conditions. | ||
| Fig. 2 1H-NMR of CA and CAAE, water peak at ∂ = 3.33 ppm has been removed from these spectra. | ||
1H-NMR results suggests that a selective reaction occurs without affecting the aliphatic hydroxyl group or the Cα–Cβ double bond.
13C-NMR analysis was also performed for further characterization and future reference, Fig. 3. The spectra of CA correspond very well with assignments found in literature.38 The product 13C-NMR reveal three newly formed peaks at ∂ = 68.95 ppm, ∂ = 117.45 ppm and ∂ = 133.87 ppm belonging to the newly formed allyl ether strengthening the claim that a selective allylation have occurred.
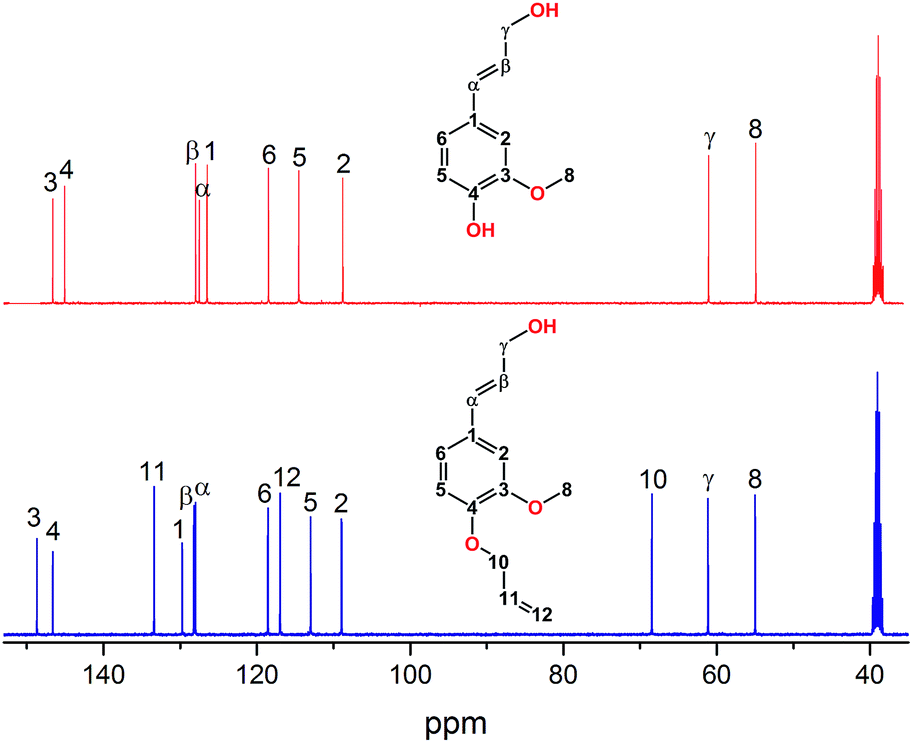 | ||
| Fig. 3 13C-NMR of CA and CAAE. | ||
2D HSQC NMR was also performed to determine and verify the structure of the product, spectrum is found in ESI Fig. S1.† The cross peak references for α, β, γ and the aromatics are found at δαH/δαC = 6.48/128.91 ppm, δβH/δβC = 6.27/129.07 ppm, δγH/δγC = 4.11/62.11 ppm, δ2H/δ2C = 7.06/109.86 ppm, δ5H/δ5C = 6.89/113.87 ppm, δ6H/δ6C = 6.89/119.48 ppm. The allyl group is found at δ10H/δ10C = 4.52/69.57 ppm, δ11H/δ11C = 6.03/134.4 ppm, δ12H/δ12C = 5.24/118.02 ppm, and δ13H/δ13C = 5.38/117.92 ppm.
The purity and molecular weight of CAAE was further analysed by GC-MS, details are found in the ESI.†
FT-IR & Raman
FT-IR and FT-Raman measurements of the CA and CAAE provide complementary data on different structural features. Spectra of the two compounds are very useful both to confirm the results of the present study but will also be of great value when using the CAAE in further modifications. Functional groups such as phenols and alcohols have strong signals in FT-IR while peaks coupled to carbon–carbon double bonds are stronger in the FT-Raman spectra. It is for example seen in the FT-IR spectra, Fig. 4, that the hydroxyl peak around 3300 cm−1 representing both the aliphatic alcohol and the phenol in CA changes to a more well-defined, although broad, peak in the spectrum for CAAE. | ||
| Fig. 4 FT-IR in the range between 2600 and 3700 cm−1. | ||
As can be seen in FT-Raman spectra, Fig. 5, the conjugated double bond between the α and β carbons is found at 1657 cm−1 in both the reagent as well as in the product. It can also be realised that a peak at 1651 cm−1 is present in the product spectra but absent in the CA, indicating the addition of a new allyl double bond. The peaks around 1600 cm−1 correspond to the aromatic vibrations of the rings.
 | ||
| Fig. 5 FT-Raman in the range between 1680 and 1570 cm−1. | ||
Conversion as a function of time
Samples from the reaction mixture was retrieved at specific intervals and analysed with NMR to determine the conversion over time. The specimens were cooled, the solvent evaporated and the dried residue re-dissolved in d6-DMSO before analysis. This procedure results in samples where the phenols are present in their deprotonated state. When CA is deprotonated several peaks shifts in the 1H-NMR spectra. The peak corresponding to the methoxy group at 3.78 ppm shifts downwards compared to the protonated CA methoxy peak. This allows for calculation of conversion through peak integrals using the deprotonated CA methoxy peak and the methoxy peak corresponding to the CAAE structure. For the same purpose the peak at 4.07 ppm belonging to the two protons on the γ carbon can be used, both methoxy as well as γ peaks are represented in Fig. 6. | ||
| Fig. 6 Utilizing integral calculation of the peaks the conversion can be achieved. Here, 55 °C after 6 hours. | ||
To give as accurate description as possible, the conversion was calculated using both the peaks corresponding to the methoxy groups as well as the γ-protons. The mean values are presented in Fig. 7 (and Table S1†). It can be seen that the conversion is well above 90% after 24 hours which really emphasis the selectivity of this reaction.
 | ||
| Fig. 7 Conversion calculated from NMR integrals, reaction temperature 55 °C. | ||
Effect of reaction temperature
In Fig. 8, the conversion achieved after 6 and 24 hours were calculated in the same way as above and shown for the different reaction temperatures; 25, 35, 45, 55, 65, 75, and 85 °C. It can clearly be seen that temperature has a great influence over the reaction rate. At 85 °C however a slight drop in conversion is observed compared to the lower temperature. 85 °C is well above the boiling point of both allyl chloride and ethanol and this result in a possible loss of reagent from the reaction vessel through the septum used. It should however be noted that a conversion of +98% could be obtained under optimal reaction conditions which is close to the detection limit of residual CA in the system. Another observation is that the reaction was performed in ethanol as a solvent providing a high concentration of primary aliphatic alcohols. The reactivity of allyl chloride towards aliphatic alcohols under the present reaction conditions must thus be minimal. The chloride represent a suitable reactivity as a leaving group in relation to the phenolate as a nucleophile. The possibility to use ethanol as a solvent for these reactions opens up new opportunities since ethanol is renewable, abundant and exhibit very good solvent properties. The used ethanol also contained 4% water demonstrating that water can be present without having any significant effects on the reaction. We can thus conclude that the reaction indeed is very selective, relatively fast, and goes to almost quantitative conversion under ambient atmosphere.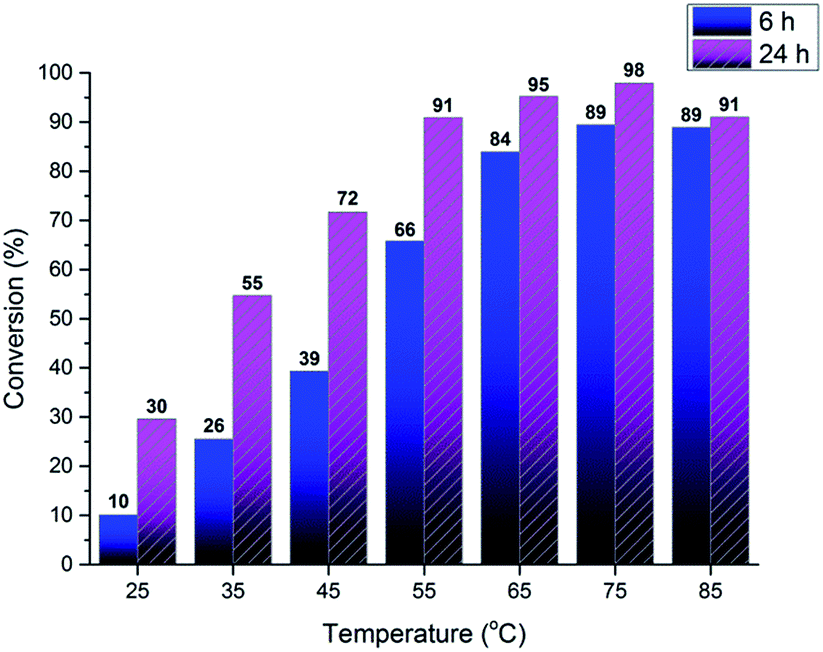 | ||
| Fig. 8 Conversion vs. temperature for 6 & 24 hour reactions. | ||
Thermal properties
Thermal analysis provides useful information on phase transitions but also about the thermal stability of the different chemical entities. It can be seen in Fig. 9 that the allylation reduced the melting temperature of from 76 °C for CA to 60 °C for CAAE. The crystallization temperature also shifted from 53 to 14 °C. An exotherm can furthermore be observed for the allylated species with a minimum at 233 °C suggesting a reaction occurring for CAAE. This analysis also provide practical information about synthesis and processing limitations since the newly formed monomer is shown to be stable up to at least 160 °C under inert conditions. | ||
| Fig. 9 DSC thermograms of the second heating scan. Samples heated from −30 to 300 °C. Both CAAE and CA are present in the graph for comparison. | ||
2D HSQC NMR analyses were performed on CAAE before and after heating to 300 °C in the DSC, Fig. S1 and S2.† The samples retrieved from the DSC were difficult to dissolve in DMSO however some useful information was obtained. Proton–carbon correlation of the allyl ether can be identified and found in both the monomer as well as in the thermally formed product. The signals have however shifted suggesting that the thermal treatment affected the allyl group. One other thing easily determined is that the H–C5 correlation of the aromatic ring is no longer present suggesting that a Claisen re-arrangement have occurred as a main reaction. During the Claisen re-arrangement the phenolic group is reformed, now with an allyl group ortho to the phenol,21 mechanism represented in Fig. S3.† Depending on application and material preferences it is thus crucial to have an understanding of this aspect when this compound is subjected to elevated temperatures. Further extensive work is needed to find the full mechanisms of these thermally driven reactions as well as the complete structure of the formed products including the insoluble residues.
Crosslinking through free radical thiol–ene coupling
The free radical thiol–ene reaction is a versatile synthesis route towards crosslinking of a wide variety of alkene monomers.39 The well documented thiol–ene reaction is one straight forward way to show the practicality of the modification and could be an interesting subject for lignin utilization in materials in the future.40,41 Crosslinked films using CAAE were made using TMP as a trifunctional thiol crosslinker in either equimolar thiol/alkene or ±10% thiol ratio. The thermoset films were analysed using FT-Raman, FT-IR and DSC to determine the final structure as well as the thermal properties of the material.In FT-Raman spectra, Fig. 10, one can clearly see the disappearance of the thiol peak around 2578 cm−1 as well as the ene peaks around 1655 cm−1. Only very small differences in the FTIR or FT-Raman spectra were seen on the samples having either 10% thiol or ene excess making a quantitative analysis of residual thiols or alkenes impossible. The FTIR and FT-Raman spectra thus confirm that the thiol–ene reaction is the dominating crosslinking reaction for this monomer and that full conversion of the both the double bonds and the thiols have been achieved.
 | ||
| Fig. 10 FT-Raman spectra of uncured CAAE and TMP mixture and a crosslinked film. Thiol peak (2578 cm−1) and ene (1655 cm−1) peak has decreased and disappeared during curing. | ||
The thermal properties of the material was determined using DSC. The Tg of the crosslinked film with an equimolar ratio thiol/alkene was determined to 17 °C upon heating and 13 °C upon cooling. No crystallinity was observed in the thermograms. The relatively low observed Tg is due to the flexible thio ether bonds formed during curing which is a well known feature for thiol–ene systems.42 Aromatics on the other hand should increase the Tg transition suggesting that larger lignin structures would give higher Tg's in this type of systems.8 The off stoichiometry based thermoset films exhibited slightly lower Tg's (around 13 °C upon heating) indicating the presence of non-reacted excess functional groups. This is also in accordance with the statement that the thiol–ene reaction is dominating in this system. The plotted DSC curves can be found in Fig. S7–S9.†
Conclusions
The present paper demonstrates a versatile route for selective modification of lignin phenols to form aryl allyl ethers. It is shown that a selective modification of the phenol in coniferyl alcohol to form coniferyl alcohol allyl ether can be performed in quantitative yields, using allyl chloride in the presence of aliphatic alcohols, alkenes, and water under aerobic conditions. Renewable, environmentally friendly protic solvents, such as ethanol, can be used without affecting the reaction. It was also determined that the reaction rate is increased when the temperature is increased without reduced selectivity of the reaction.It was furthermore demonstrated that the formed product, CAAE, is stable up to at least 160 °C, in inert conditions, thus allowing for further chemical transformations to be made. Claisen re-arrangement and other reactions start to occur at temperatures above 175 °C. In addition to the selective modification it was also proven that CAAE can be used as a monomer in thermosets using free radical thiol–ene polymerization as the crosslinking reaction. Thermoset films with a full conversion of the double bonds could readily be obtained using this polymerization route.
Acknowledgements
The authors are grateful to the Knut and Alice Wallenberg Foundation for financial support through the Wallenberg Wood Science Center at KTH Royal Institute of Technology.References
- L. Y. Ljungberg, Mater. Des., 2007, 28, 466–479 CrossRef.
- G. Gellerstedt and G. Henriksson, Lignins: major sources, structure and properties in Monomers, Polymers and Composites from Renewable Resources, ed. M. Belgacem and A. Gandini, Elsevier, 1st edn, 2008, Ch. 9 Search PubMed.
- M. Fache, E. Darroman, V. Besse, R. Auvergne, S. Caillol and B. Boutevin, Green Chem., 2014, 16, 1987–1998 RSC.
- H. A. Meylemans, B. G. Harvey, J. T. Reams, A. J. Guenthner, L. R. Cambrea, T. J. Groshens, L. C. Baldwin, M. D. Garrison and J. M. Mabry, Biomacromolecules, 2013, 14, 771–780 CrossRef CAS PubMed.
- B. G. Harvey, A. J. Guenthner, H. A. Meylemans, S. R. L. Haines, K. R. Lamison, T. J. Groshens, L. R. Cambrea, M. C. Davis and W. W. Lai, Green Chem., 2015, 17, 1249–1258 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 4862–4887 RSC.
- S. E. Lebo, J. D. Gargulak and T. J. McNally, in Kirk–Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2000, DOI:10.1002/0471238961.12090714120914.a01.pub2.
- P. Dournel, E. Randrianalimanana, A. Deffieux and M. Fontanille, Eur. Polym. J., 1988, 24, 843–847 CrossRef CAS.
- G. Gellerstedt, Ind. Crops Prod., 2015, 77, 845–854 CrossRef CAS.
- P. Tomani, Cellul. Chem. Technol., 2010, 44, 53–58 CAS.
- F. V. Antoine Duval, C. Crestini and M. Lawoko, Holzforschung, 2015, 70, 11–20 Search PubMed.
- C. Crestini, F. Melone, M. Sette and R. Saladino, Biomacromolecules, 2011, 12, 3928–3935 CrossRef CAS PubMed.
- R. Auvergne, S. Caillol, G. David, B. Boutevin and J.-P. Pascault, Chem. Rev., 2014, 114, 1082–1115 CrossRef CAS PubMed.
- G. Gellerstedt and G. Henriksson, in Monomers, Polymers and Composites From Renewable Resources, ed. M. N. B. Gandini, Elsevier, Amsterdam, 2008, pp. 201–224, DOI:10.1016/B978-0-08-045316-3.00009-0.
- J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. F. Schatz, J. M. Marita, R. D. Hatfield, S. A. Ralph, J. H. Christensen and W. Boerjan, Phytochem. Rev., 2004, 3, 29–60 CrossRef CAS.
- J. J. Meister, J. Macromol. Sci., Polym. Rev., 2002, 42, 235–289 CrossRef.
- P. P. Klemchuk and P.-L. Horng, Polym. Degrad. Stab., 1991, 34, 333–346 CrossRef CAS.
- I. Volf, I. Ignat, M. Neamtu and V. I. Popa, Chem. Pap., 2013, 68, 121–129 Search PubMed.
- L. Zoia, A. Salanti, P. Frigerio and M. Orlandi, BioResources, 2014, 9, 6540–6561 CrossRef CAS.
- L. C. Over and M. A. R. Meier, Green Chem., 2015, 18, 197–207 RSC.
- J. Xin, P. Zhang, K. Huang and J. Zhang, RSC Adv., 2014, 4, 8525–8532 RSC.
- T. Wertheim, Justus Liebigs Ann. Chem., 1844, 51, 289 CrossRef.
- E. S. Jensen, P. Gatenholm, S. R. Nanguneri and L. J. Mathias, J. Appl. Polym. Sci., 1991, 42, 2681–2689 CrossRef CAS.
- H. U. Pohl, H.-J. Traenckner and H. J. Rosenkranz, Angew. Makromol. Chem., 1981, 98, 1–34 CrossRef CAS.
- J. v. Braun and R. Murjahn, Ber. Dtsch. Chem. Ges., 1926, 59, 1202–1209 CrossRef.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- M. V. Walter, P. Lundberg, A. Hult and M. Malkoch, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2990–2995 CrossRef CAS.
- I. Carlsson, A. Harden, S. Lundmark, A. Manea, N. Rehnberg and L. Svensson, in Photoinitiated Polymerization, American Chemical Society, 2003, vol. 847, ch. 6, pp. 65–75 Search PubMed.
- H. Yuan, X. Lu, Z. Zeng, J. Yang and Y. Chen, J. Appl. Polym. Sci., 2004, 92, 2765–2770 CrossRef CAS.
- T. J. Prosser, J. Am. Chem. Soc., 1961, 83, 1701–1704 CrossRef CAS.
- X. Coqueret and G. Wegner, Organometallics, 1991, 10, 3139–3145 CrossRef CAS.
- F. Lu and J. Ralph, J. Agric. Food Chem., 1998, 46, 1794–1796 CrossRef CAS.
- H. Kim and J. Ralph, J. Agric. Food Chem., 2005, 53, 3693–3695 CrossRef CAS PubMed.
- P. Werle, M. Morawietz, S. Lundmark, K. Sörensen, E. Karvinen and J. Lehtonen, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, DOI:10.1002/14356007.a01_305.pub2.
- S. Ralph, J. Ralph, L. Landucci and L. Landucci, US Forest Prod. Lab., Madison, WI, http://ars.usda.gov/Services/docs.html, 2004.
- A. F. Jacobine, Thiol–ene photopolymers, Elsevier, London, UK, 1993 Search PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS PubMed.
- Q. Li, H. Zhou, D. A. Wicks and C. E. Hoyle, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5103–5111 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 2D HSQC NMR, full FT-IR and FT-Raman spectra, and tables of conversions. See DOI: 10.1039/c6ra21447a |
| This journal is © The Royal Society of Chemistry 2016 |