
An easily accessible and recyclable copper nanoparticle catalyst for the solvent-free synthesis of dipyrromethanes and aromatic amines†
Sengan Megarajan,
Khan Behlol Ayaz Ahmed,
Rajamani Rajmohan,
Pothiappan Vairaprakash * and
Veerappan Anbazhagan*
* and
Veerappan Anbazhagan*
Department of Chemistry, School of Chemical and Biotechnology, SASTRA University, Thanjavur, Tamil Nadu, India. E-mail: vairaprakash@scbt.sastra.edu; anbazhagan@scbt.sastra.edu; anbugv@gmail.com
First published on 20th October 2016
Abstract
A facile method to prepare a reusable copper nanocatalyst is reported. Transmission electron microscopy shows that the CuNPs are spherical in nature and reside in the nanoscopic template of the capping agent, guar gum. Powder X-ray diffraction studies confirm the crystalline nature of the CuNPs. The catalytic efficiency and recyclability of the synthesized CuNPs were demonstrated through the synthesis of dipyrromethanes (DPMs) and aromatic amines. Various DPMs are obtained in very good yields under solvent-free conditions and nitroarenes are converted to the corresponding amino compounds within 10 min, as evidenced by the kinetic study.
Introduction
The physicochemical nature of a material gets modified when translated into nanodimension and offers improved physical, chemical and biological activities. For example, gold is considered an inert metal in its bulk form, while at the same time nanogold exhibits excellent catalytic activity in several organic transformations.1 Nanocatalysts are highly advantageous compared to regular catalysts because they provide an increased catalytic surface to volume ratio, thereby minimizing the catalytic loading. Of late, the catalytic activities of copper nanoparticles (CuNPs) have been explored in Ullmann reactions, Heck reactions, Sonogashira reactions,1 dipolar cycloaddition, Huisgen [3 + 2]-cycloaddition and so forth.2 Economical and eco-friendly access and reusability facilitates the adaptability of this material in industry. Herein, we report an easy method to synthesize CuNPs using a biopolymer, guar gum, as a capping agent. The choice of guar gum was based on its benign nature and the fact that it is easy to obtain from renewable resources.3 The synthesized CuNPs were characterized by transmission electron microscopy, powder X-ray diffraction and energy dispersive X-ray spectroscopy. The effectiveness and reusability of the catalyst were demonstrated through the synthesis of dipyrromethanes and aromatic amines. Dipyrromethanes (DPMs) are versatile key intermediates in the construction of various functional organic molecules such as BODIPY dyes,4 porphyrins,5 chlorins,6 corroles7 etc. for photonics/electronics applications including: (i) photo-induced electron transfer,8 (ii) molecular switches,9 (iii) spin switching,10 (iv) triplet sensitized conjugated polymers,11 (v) dyes,12 (vi) bio-imaging,13 (vii) photodynamic therapy,14 (viii) electrocatalytic oxygen reduction,15 (ix) sensors,16 (x) fluorescent probes17 etc. Traditionally, DPMs are synthesized by the reaction of pyrrole with aldehydes, catalyzed by Lewis or Brønsted acids including HCl, CF3COOH, InCl3, I2/AcOH, CAN, BF3–etherate, TsOH and cation exchange resins.18 These reactions do not simply stop at the dipyrromethane stage; they proceed to form oligomeric/polymeric compounds, necessitating tedious purification processes and also resulting in low yields. To avoid the formation of polymeric byproducts and to selectively stop the reaction at the dipyrromethane stage, several modifications have been introduced in to the synthesis, such as (i) employing 2,4-substituted pyrrole19 and (ii) a solvent-free synthesis employing pyrrole as the reagent and solvent.20,21 The former case is only applicable to the synthesis of BODIPY dyes, as substituted DPMs were obtained and these substituted DPMs cannot be converted into tetra-pyrrolic macrocycles. In the latter case, the use of excess pyrrole limits the versatility of this methodology. In these reactions, the acidity of the catalyst has a significant impact on the reaction outcome and selectivity.21 Among the catalysts reported for DPM synthesis, promising results were obtained in reactions employing a more expensive InCl3 catalyst and 100 equivalents of pyrrole.21 The milder acidity of InCl3 resulted in the selective formation of DPM.21 Recently, we have disclosed the synthesis of dipyrromethane analogs from fructose derived HMF using celite supported InCl3 as a recyclable catalyst.22 Herein we report simple, easily accessible and recyclable copper nanoparticles for DPM synthesis. These CuNPs provide more selectivity in DPM synthesis. In addition to DPM synthesis, our catalytic system is found to be more efficient in reducing aromatic nitro compounds to the corresponding amines.The reduction of aromatic nitro compounds to the corresponding amine is an active area of research, as amines and their derivatives are important starting materials for the synthesis of chemicals including dyes, pharmaceuticals, and anticorrosive lubricants.22 In light of the commercial importance, many methods have been developed for the production of aromatic amines which include the catalytic hydrogenation of aromatic nitro compounds with hydrogen gas in the presence of Pd, Pt, Ni and Rh metal.23 In recent years, instead of H2 gas, sodium borohydride has been used as a hydrogen source with metal nanoparticles as the catalysts.24 Among the available methods for the synthesis of aromatic amines, nitroarene reduction using NaBH4/metal nanoparticles is considered a trusted model reaction to explore the catalytic activity of nanoparticles.25 In this work, we exploited the chromophoric nature of the aromatic NO2 group and determined the rate of reduction using absorbance spectroscopy and the results are discussed.
Results and discussion
Synthesis of copper nanoparticles
The reusable copper nanocatalyst (CuNPs) was synthesized by a bottom-up approach using the precursor copper(II) chloride and the biopolymer guar gum and hydrazine as a capping and reducing agent, respectively. The aqueous CuNPs were dehydrated by heating at 100 °C. The colloidal reddish brown solution turned to a brownish black precipitate, confirming the removal of water (Fig. 1). The obtained precipitate is insoluble in water indicating that the average strength of the biopolymer–biopolymer interaction is significantly increased compared to the water–polymer interaction. | ||
| Fig. 1 Photograph representing the preparation of the reusable CuNP catalyst. | ||
Energy dispersive X-ray spectroscopy (EDS) confirms the presence of copper in our preparation (Fig. 2A). The noticeable amount of oxygen in the EDS spectrum can be attributed to the oxygen rich capping agent. X-ray diffraction showed diffraction pattern peaks at 43.32°, 50.39° and 74.08°, which correspond to the crystal planes (111), (200) and (220), respectively (Fig. 2B). The absence of diffraction peaks corresponding to copper oxides (CuO, Cu2O) suggests that the prepared NPs are composed of metallic copper. Transmission electron microscopy (TEM) revealed that the particles are spherical nature with a size range of 100 to 300 nm. TEM images clearly show that the CuNPs reside in the nanoscopic template of the capping agent (Fig. 3). The observed ring pattern in the selected area diffraction pattern confirms the crystalline nature of the prepared CuNPs.
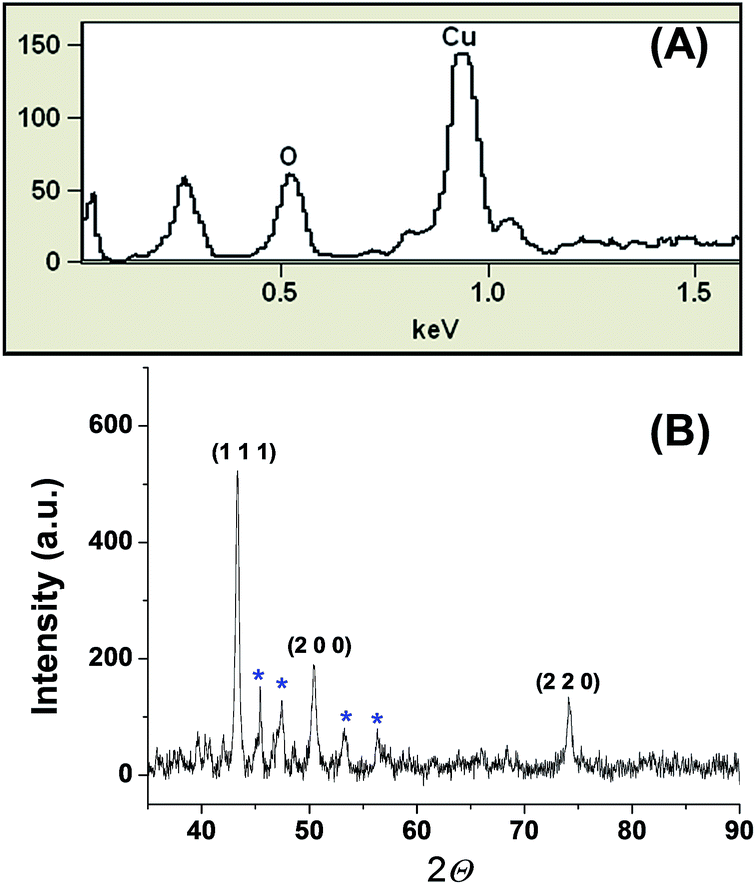 | ||
| Fig. 2 (A) EDS spectrum and (B) powder X-ray diffraction pattern of the CuNPs. The sharp peaks (blue star) can be attributed to the capping agent guar gum, which is known for its crystalline nature. | ||
 | ||
| Fig. 3 Transmission electron micrographs of the CuNPs with a scale bar of (A) 500 nm and (B) 200 nm. Inset corresponds to the SAED pattern. | ||
Synthesis of dipyrromethanes
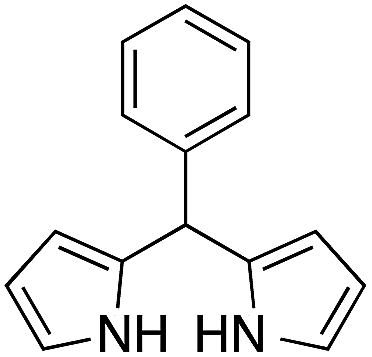
The catalytic behavior of the CuNPs was explored in the synthesis of dipyrromethanes and aromatic amines. In the synthesis of DPM, a mixture containing 4-anisaldehyde and pyrrole (10 equiv.) was treated with a suspension of CuNPs in water at 30 °C. Under these conditions, the aldehyde was consumed completely within 8 h. Analysis of the reaction mixture showed the formation of more byproducts and DPM was obtained in very low yield (∼5%, Table 1, entry 1). When the reaction was repeated under solvent-free conditions using 10 equiv. of pyrrole, complete consumption of the aldehyde was observed after 15 h, with very good selectivity towards the product and the corresponding DPM was obtained in 63% isolated yield (Table 1, entry 2). Formation of DPM in trace quantities in an experiment carried out without CuNPs evidently indicated the role of the CuNPs as a catalyst in DPM synthesis (Table 1, entry 3). The catalytic behavior of the CuNPs is attributed to the increase in nucleophilicity of pyrrole upon adsorption over CuNPs.| Entry | Reactant | Product | Time (h) | Yield (%) |
|---|---|---|---|---|
| a A mixture of aldehyde (0.5 mmol), pyrrole (5 mmol, 10 equiv.) and copper nanoparticles (5 mg) was stirred at 30 °C. After the completion of the reaction, the reaction mixture was diluted with dichloromethane (5 mL), centrifuged and the catalyst was recovered as the residual fraction. The supernatant was purified by column chromatography.b Reaction was carried out in water.c Reaction without CuNPs.d Recycled catalyst (II cycle).e Recycled catalyst (III cycle).f Recycled catalyst (IV cycle).g A mixture of 3-nitrobenzaldehyde (0.5 mmol), 2,4-dimethylpyrrole (5 mmol, 10 equiv.) and copper nanoparticles (5 mg) was stirred at 30 °C.h Experiment was carried out with 3 equiv. of 2,4-dimethylpyrrole. | ||||
| 1b | 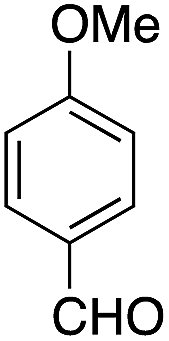 |
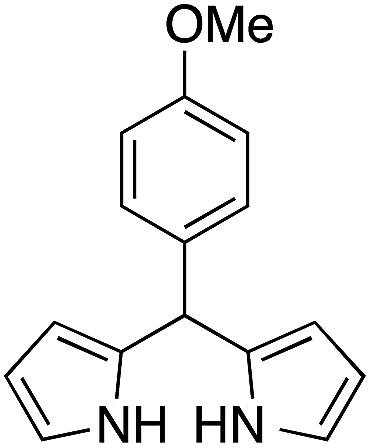 |
8 | 5 |
| 2 | 15 | 63 | ||
| 3c | 24 | Trace | ||
| 4 |  |
 |
15 | 65 |
| 5 | 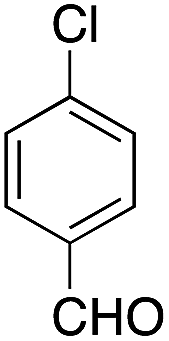 |
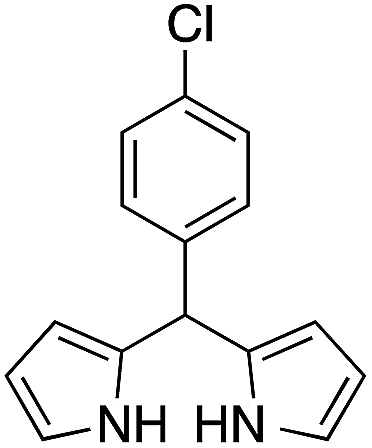 |
15 | 71 |
| 6 |  |
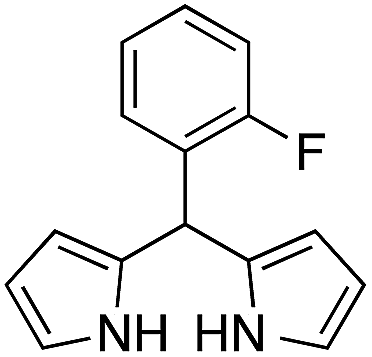 |
15 | 63 |
| 7 | 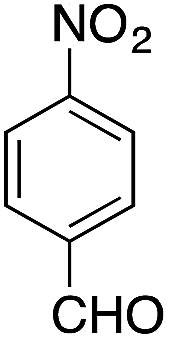 |
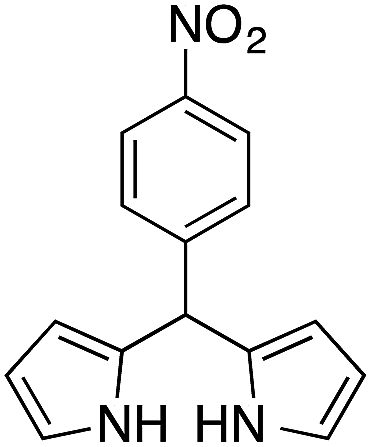 |
12 | 75 |
| 8 | 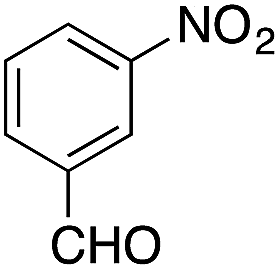 |
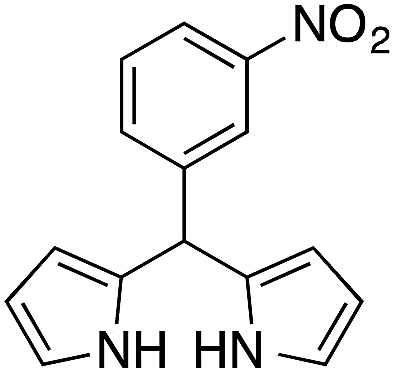 |
12 | 77 |
| 9d | 12 | 73 | ||
| 10e | 12 | 71 | ||
| 11f | 12 | 74 | ||
| 12g | 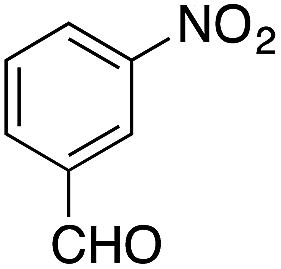 |
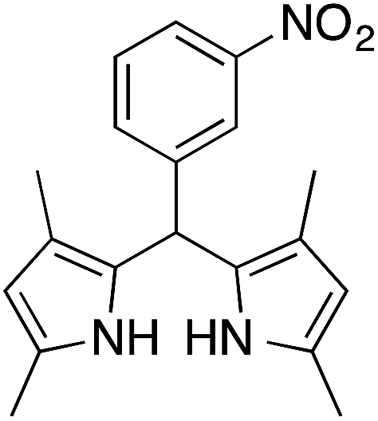 |
0.5 | 75 |
| 13h | 0.5 | 72 | ||
| 14 | Acetophenone | — | 24 | — |
| 15 | Cyclohexanone | — | 24 | — |
On adsorption over CuNPs, the nucleophilicity of pyrrole is increased and it reacts with the electrophilic carbon of the aldehyde. The resulting intermediate reacts with another pyrrole adsorbed on the CuNPs to yield dipyrromethane after removal of the water molecule. The reactivities of other aryl aldehydes were tested at this temperature (Table 1, entries 4–8). Electron withdrawing substituents present in the aldehyde facilitated better conversion to DPM. Among various aldehydes, the better conversion was obtained with 3-nitrobenzaldehyde and the product was obtained in a 77% yield (Table 1, entry 8). In all experiments, the catalyst was recovered by diluting the reaction mixture with dichloromethane (5 mL), followed by centrifugation for 15 min and then the catalyst was recovered as a residual fraction. The recovered catalyst showed similar catalytic activity without any deterioration in yields after up to 4 cycles (Table 1, entries 8–11). In an experiment using 2,4-dimethylpyrrole, 3-nitrobenzaldehyde was consumed within 30 min and the corresponding DPM was obtained in 75% yield (Table 1, entry 12). Use of 2,4-dimethylpyrrole in a stoichiometric amount (3 equiv.) did not reduce the product yield (Table 1, entry 13). In the copper nanoparticle catalyzed reaction of pyrrole with aromatic and aliphatic ketones, the corresponding dipyrromethanes were not obtained even after 24 h stirring at room temperature (Table 1, entries 14 and 15).
Nitroarene reduction kinetics
In the synthesis of aromatic amines, nitroarenes can react with NaBH4 in the presence of CuNPs at room temperature. Under these conditions, nitroarenes were converted to the corresponding amines within 10 min. Taking advantage of the chromophoric nature of the nitroaromatics, the reaction was followed by UV-vis spectroscopy. During the subsequent addition of the CuNPs, the absorbance peak at 400 nm disappears with the appearance of a new peak at 300 nm, indicating the conversion of nitrophenol to aminophenol. In a typical reaction of 4-nitrophenol, the addition of NaBH4 induces a bathochromic shift from 317 nm to 400 nm due to the formation of the nitrophenolate ion (Fig. 4).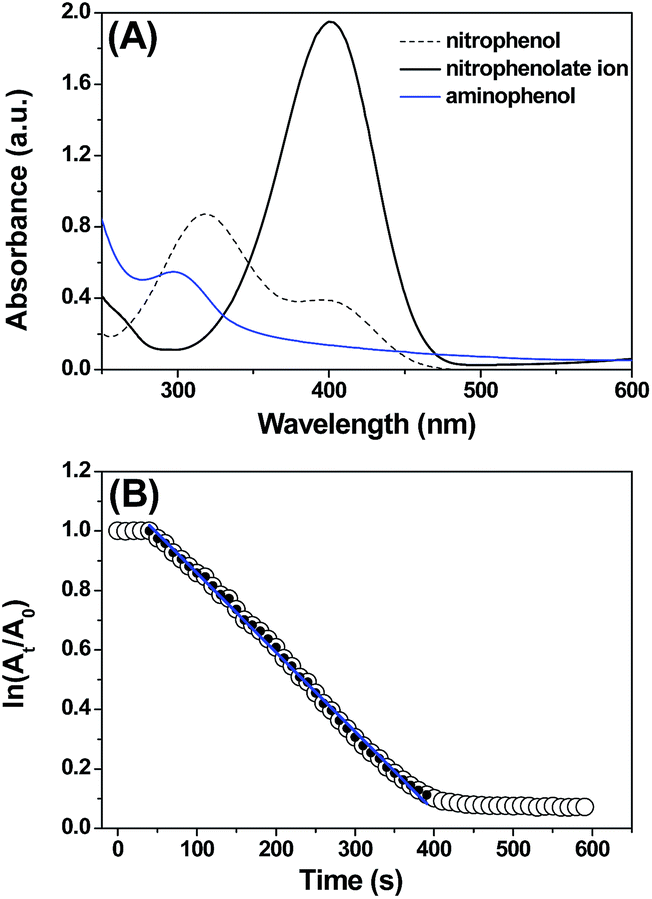 | ||
| Fig. 4 (A) UV-vis spectra for the reduction of nitrophenol to aminophenol in the presence of the CuNP catalyst and NaBH4 in water (100 μM nitrophenol, 1 mg CuNPs, 20 mg NaBH4). (B) Time-dependent absorption of the nitrophenolate ion at 400 nm, with an induction period of t0. The line corresponds to the linear section, from which k was determined. | ||
Interestingly, neither NaBH4 nor the CuNPs alone reduces the nitroaromatics, suggesting that the CuNPs and NaBH4 serve as the catalyst and reducing agent, respectively, and that both are required for this chemical transformation. The change in the absorption peak during the progress of the reduction reaction allowed us to determine the kinetics of the reduction process using absorption spectroscopy. The kinetics experiment was initiated by addition of NaBH4 and CuNPs to the nitrophenol aqueous solution and was followed by the time-dependent change in the absorbance at 400 nm. Initially, an induction time (t0) was observed, which is typical for a heterogeneous catalytic process and is commonly attributed to activation or restructuring of the metal surface by the substrate before initiating the reaction.26
After t0, the reduction reaction starts as evidenced by the decrease in the absorbance at 400 nm and stops changing after all of the substrate is converted to the product (Fig. 4A). In this catalytic reaction, the concentration of NaBH4 was much higher than that of the substrate. Therefore, the reduction kinetics can be described by the pseudo first-order rate law: ln(At/A0) = −kt, where k is the apparent first-order rate constant and t is the reaction time. At and A0 are the absorbance of the substrate at time t and 0, respectively. Fig. 4B shows the plot of ln(At/A0) versus reaction time for the reduction of nitrophenol. The rate constant, k was obtained directly from the slope of the linear part of the kinetics trace and is presented in Table 2. Nitroarene reduction was performed through IV steps as described in Fig. 5.26 In the first step, hydrogen released from NaBH4 gets adsorbed onto the nanoparticle surfaces followed by adsorption of nitroarenes onto the nanoparticle surfaces through the nitro group (step II). Then, the nitroarenes on the nanoparticle surfaces were reduced by the adsorbed hydrogen to aromatic amines. The scope of the catalytic system was tested with several other nitroaromatic compounds (Table 2). Formation of the amine is confirmed by 1H NMR spectroscopic analysis of the resulting crude mixture (Table 2, entries 2 & 8; Fig. S20 & S21†).
| Entry | Reactant | Product | Induction time (s) | k (s−1) |
|---|---|---|---|---|
| a NaBH4 (20 mg) and copper nanoparticles (1 mg) were added to 3 mL of an aqueous solution/suspension of the nitrocompound (0.1 mmol) which was followed by the absorption spectra changes with respect to time. Kinetics traces were fitted with the pseudo first-order equation and the k value is reported.b Using fresh catalyst.c Using recycled catalyst (II cycle).d Using recycled catalyst (III cycle).e Using recycled catalyst (IV cycle).f Using recycled catalyst (V cycle).g No appreciable change in the absorbance spectrum on nitro-reduction, as monitored by TLC analysis and confirmed by 1H NMR spectroscopy. | ||||
| 1 | 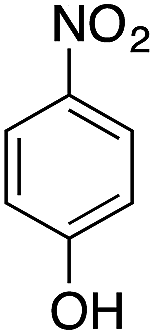 |
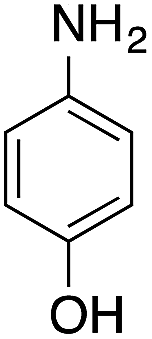 |
40.2 | 0.00267b |
| 0.00198c | ||||
| 0.00288d | ||||
| 0.00258e | ||||
| 0.00199f | ||||
| 2 | 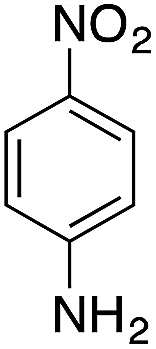 |
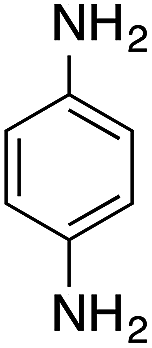 |
70.2 | 0.00343 |
| 3 | 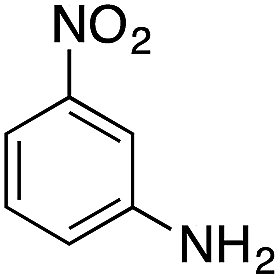 |
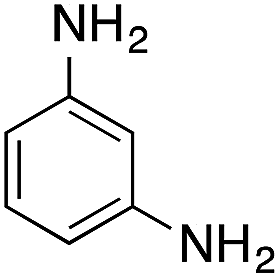 |
79.8 | 0.00077 |
| 4 | 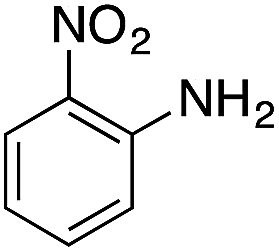 |
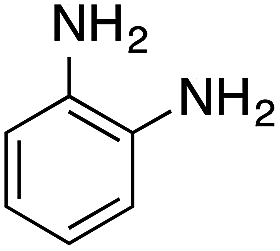 |
110.4 | 0.00211 |
| 5 | 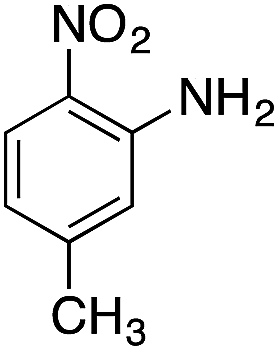 |
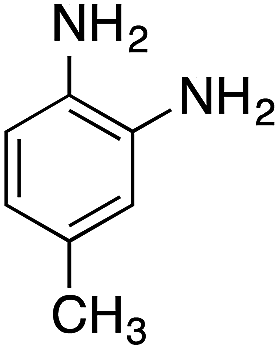 |
110.4 | 0.00237 |
| 6 | 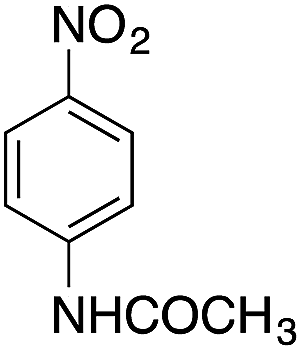 |
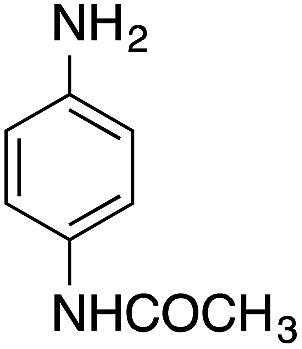 |
170.4 | 0.00659 |
| 7 | 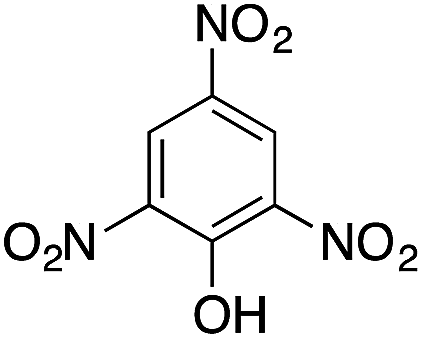 |
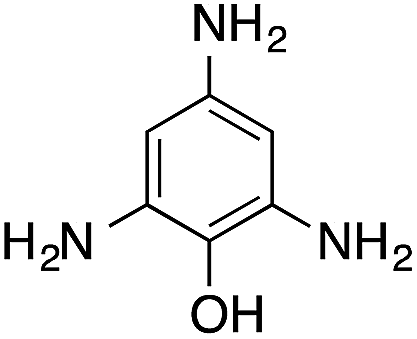 |
110.4 | 0.00236 |
| 8g | 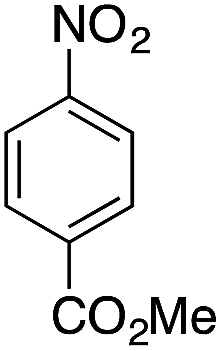 |
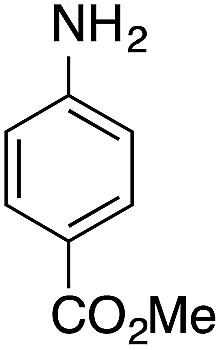 |
— | — |
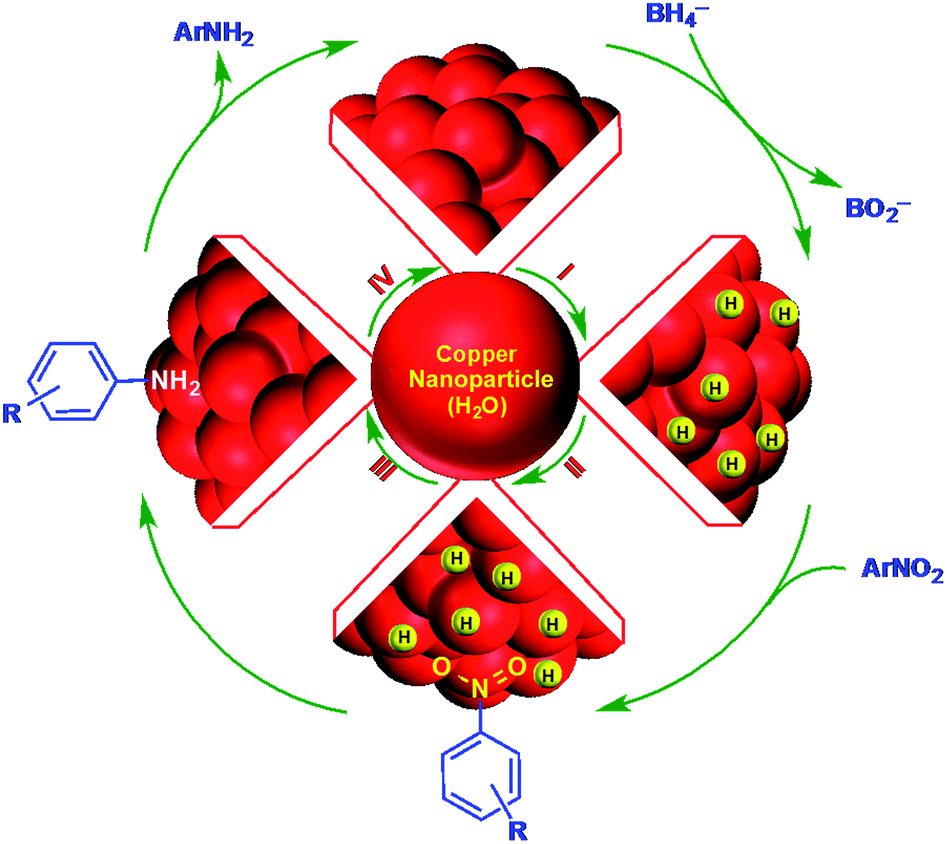 | ||
| Fig. 5 Nitro-reduction over CuNPs using NaBH4. | ||
In all experiments, the catalyst was recovered by centrifugation and the residue was subjected to an acetone wash prior to use in the next cycle. The reusability of the catalyst was studied with the nitrophenol system. The recovered catalyst efficiently converted nitrophenol to aminophenol for up to five cycles as evidenced by the similar rate constants (10−3 s−1). The XRD and TEM analysis of the recovered catalyst after the 5th cycle confirmed that there is not a considerable change in the morphology of the catalyst (Fig. S18†). As noted from Table 2, irrespective of the substituent, all the tested nitro-aromatic compounds show a rate constant in the order of 10−3 s−1, which is consistent with those of substrate-supported metallic nanoparticle catalyzed reactions reported in the literature.27 The difference in the induction time observed for the different substrates can be attributed to the steric hindrance, which may delay the activation or restructuring of the substrate on the metal surface. Nevertheless, the procedure described here makes it simple to prepare amino compounds from nitroarenes in a shorter time. With our recyclable and easily accessible copper catalyst system, we have obtained rate constant values for nitro reduction reactions comparable with those of previously reported nanocatalytic systems (Table 3).28
Conclusions
In summary, we have developed easy access to dry copper nanoparticles, with retained morphology of the solution state. The isolated nanoparticles have been tested in the synthesis of dipyrromethanes and aromatic amines. In the synthesis of DPMs, the nucleophilicity of pyrrole is enhanced on adsorption over the CuNPs and it reacts at the electrophilic site of the aldehyde. The corresponding DPMs were obtained in very good yields. In addition, the catalytic efficiency of the CuNPs was investigated with a well-known 4-nitrophenol reduction kinetics experiment which was extended to nitroarenes. Irrespective of the substituent, all the tested nitroarenes got reduced to amino compounds in less than 10 min. The recyclability of the CuNPs was exhibited evidently in both the syntheses. These findings on easy access to CuNPs and their use in the synthesis of dipyrromethanes and aromatic amines will provide a boost for the exploration of applications of simple to access CuNPs in various organic syntheses.Experimental
General information
1H NMR (300 MHz) spectra were recorded on a Bruker-300-AVANCE II spectrometer, with chloroform-d as a solvent and tetramethylsilane (TMS) as a reference (δ = 0 ppm). The chemical shifts are expressed in δ downfield from the signal of the internal TMS. All yields reported are of isolated materials judged to be homogeneous by TLC and NMR spectroscopy. The size, morphology and crystallinity were characterized by high-resolution transmission electron microscopy (HR-TEM) (JEOL-JEM 1011, Japan) with an accelerating voltage of 200 kV. Samples for the TEM measurements were prepared by placing a drop of NP solution onto a graphite grid and drying it in a vacuum.To calculate the rate constant, the obtained kinetics trace was analyzed by the pseudo first-order rate law:
| ln(At/A0) = −kt | (1) |
Acknowledgements
PV (SB/FT/CS-003/2014) and VA (SB/FT/LS-217/2012) thank the Department of Science and Technology, Government of India for financial support. P. V. thanks SASTRA University, Thanjavur for the Prof. T. R. Rajagopalan research grant. The central research facility (R&M/0021/SCBT-007/2012-13), SASTRA University is acknowledged for the infrastructure.Notes and references
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 122, 4469–4509 CrossRef PubMed.
- M. B. Gawande, A. Goswami, F. X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS PubMed.
- D. Das, T. Ara, S. Dutta and A. Mukherjee, Bioresour. Technol., 2011, 102, 5878–5883 CrossRef CAS PubMed.
- (a) A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC; (b) J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- (a) Z. Yao, J. Bhaumik, S. Dhanalekshmi, M. Ptaszek, P. A. Rodriguez and J. S. Lindsey, Tetrahedron, 2007, 63, 10657–10670 CrossRef CAS PubMed; (b) T. Sahin, P. Vairaprakash, K. E. Borbas, T. Balasubramanian and J. S. Lindsey, J. Porphyrins Phthalocyanines, 2015, 19, 664–678 CrossRef.
- (a) M. Krayer, M. Ptaszek, H.-J. Kim, K. R. Meneely, D. Fan, K. Secor and J. S. Lindsey, J. Org. Chem., 2010, 75, 1016–1039 CrossRef CAS PubMed; (b) K. Aravindu, O. Mass, P. Vairaprakash, J. W. Springer, E. Yang, D. M. Niedzwiedzki, C. Kirmaier, D. F. Bocian, D. Holten and J. S. Lindsey, Chem. Sci., 2013, 4, 3459–3477 RSC; (c) J. S. Lindsey, Chem. Rev., 2015, 115, 6534–6620 CrossRef CAS PubMed.
- (a) D. T. Gryko and K. J. Jadach, J. Org. Chem., 2001, 66, 4267–4275 CrossRef CAS PubMed; (b) D. T. Gryko and K. E. Piechot, J. Porphyrins Phthalocyanines, 2002, 6, 81–97 CrossRef CAS; (c) G. Aydin, B. Temelli and C. Unaleroglu, Eur. J. Org. Chem., 2015, 7583–7593 CrossRef CAS.
- T. Higashino, T. Yamada, M. Yamamoto, A. Furube, N. V. Tkachenko, T. Miura, Y. Kobori, R. Jono, K. Yamashita and H. Imahori, Angew. Chem., Int. Ed., 2016, 55, 629–633 CrossRef CAS PubMed.
- R. Arumugaperumal, V. Srinivasadesikan, R. Ramakrishnam, V. Mandapati, M.-C. Lin, T. Shukla, R. Singh and H.-C. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 26491–26503 CAS.
- M. Dommaschk, C. Naether and R. Herges, J. Org. Chem., 2015, 80, 8496–8500 CrossRef CAS PubMed.
- R. Andernach, H. Utzat, S. D. Dimitrov, I. McCulloch, M. Heeney, J. R. Durrant and H. Bronstein, J. Am. Chem. Soc., 2015, 137, 10383–10390 CrossRef CAS PubMed.
- (a) H. Zhao, J. Long, X. Luo, B. Zhao and S. Tan, Dyes Pigm., 2015, 122, 168–176 CrossRef CAS; (b) J. Liao, Y. Wang, Y. Xu, H. Zhao, X. Xiao and X. Yang, Tetrahedron, 2015, 71, 5078–5084 CrossRef CAS.
- (a) M. Jurasek, S. Rimpelova, E. Kmonickova, P. Drasar and T. Ruml, J. Med. Chem., 2014, 57, 7947–7954 CrossRef CAS PubMed; (b) R. Xiong, J. Andres, K. Scheffler and K. E. Borbas, Dalton Trans., 2015, 44, 2541–2553 RSC; (c) D. Kand, T. Saha, M. Lahiri and P. Talukdar, Org. Biomol. Chem., 2015, 13, 8163–8168 RSC.
- (a) R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC; (b) J. M. Dabrowski, L. G. Arnaut, M. M. Pereira, C. J. P. Monteiro, K. Urbanśka, S. Simões and G. Stochel, ChemMedChem, 2010, 5, 1770–1780 CrossRef CAS PubMed; (c) M. B. Ballatore, M. B. Spesia, M. E. Milanesio and E. N. Durantini, Eur. J. Med. Chem., 2014, 83, 685–694 CrossRef CAS PubMed.
- S. Amanullah, P. K. Das, S. Samanta and A. Dey, Chem. Commun., 2015, 51, 10010–10013 RSC.
- (a) A. Hafuka, R. Kando, K. Ohya, K. Yamada, S. Okabe and H. Satoh, Bull. Chem. Soc. Jpn., 2015, 88, 447–454 CrossRef CAS; (b) Z. Li, L.-J. Li, T. Sun, L. Liu and Z. Xie, Dyes Pigm., 2016, 128, 169–169 Search PubMed.
- J. C. Er, C. Leong, C. L. Teoh, Q. Yuan, P. Merchant, M. Dunn, D. Sulzer, D. Sames, A. Bhinge, D. Kim, S.-M. Kim, M.-H. Yoon, L. W. Stanton, S. H. Je, S.-W. Yun and Y.-T. Chang, Angew. Chem., Int. Ed., 2015, 54, 2442–2446 CrossRef CAS PubMed.
- (a) N. A. M. Pereira and T. M. V. D. PinhoeMelo, Org. Prep. Proced. Int., 2014, 46, 183–213 CrossRef CAS; (b) D. T. Gryko, D. Gryko and C.-H. Lee, Chem. Soc. Rev., 2012, 41, 3780–3789 RSC; (c) Y. Pareek, M. Ravikanth and T. K. Chandrashekar, Acc. Chem. Res., 2012, 45, 1801–1816 CrossRef CAS PubMed; (d) J. S. Lindsey, Acc. Chem. Res., 2010, 43, 300–311 CrossRef CAS PubMed; (e) L. Zoli and P. G. Cozzi, ChemSusChem, 2009, 2, 218–220 CrossRef CAS PubMed.
- (a) D. A. Smithen, A. E. G. Baker, M. Offman, S. M. Crawford, T. S. Cameron and A. Thompson, J. Org. Chem., 2012, 77, 3439–3453 CrossRef CAS PubMed; (b) F. E. Alemdaroglu, S. C. Alexander, D. Ji, D. K. Prusty, M. Börsch and A. Herrmann, Macromolecules, 2009, 42, 6529–6536 CrossRef CAS.
- C.-H. Lee and J. S. Lindsey, Tetrahedron, 1994, 50, 11427–11440 CrossRef CAS.
- J. K. Laha, S. Dhanalekshmi, M. Taniguchi, A. Ambroise and J. S. Lindsey, Org. Process Res. Dev., 2003, 7, 799–812 CrossRef CAS.
- R. Rajmohan, S. Gayathri and P. Vairaprakash, RSC Adv., 2015, 5, 100401–100407 RSC.
- (a) S. Panigrahi, S. Basu, S. Praharaj, S. Pande, S. Jana, A. Pal, S. K. Ghosh and T. Pal, J. Phys. Chem. C, 2007, 111, 4596–4605 CrossRef CAS; (b) M. J. Vaidya, S. M. Kulkarni, R. V. Chaudhari, C. V. Rode, M. J. Vaidya and R. V. Chaudhari, Org. Process Res. Dev., 1999, 3, 465–470 CrossRef; (c) N. Comisso, S. Cattarin, S. Fiameni, R. Gerbasi, L. Mattarozzi, M. Musiani, L. Vazquez-Gomez and E. Verlato, Electrochem. Commun., 2012, 25, 91–93 CrossRef CAS; (d) K. B. Narayanan and N. Sakthivel, J. Hazard. Mater., 2011, 189, 519–525 CrossRef CAS PubMed; (e) T. L. Lai, K. F. Yong, J. W. Yu, J. H. Chen, Y. Y. Shu and C. B. Wang, J. Hazard. Mater., 2011, 185, 366–372 CrossRef CAS PubMed; (f) D. Makovec, M. Sajko, A. Selisnik and M. Drofenik, Mater. Chem. Phys., 2011, 129, 83–89 CrossRef CAS.
- (a) P. Lara and K. Philippot, Catal. Sci. Technol., 2014, 4, 2445–2465 RSC; (b) J. Liu, J. Cui, F. Vilela, J. He, M. Zeller, A. D. Hunter and Z. Xu, Chem. Commun., 2015, 51, 12197–12200 RSC; (c) W.-G. Jia, Y.-C. Dai, H.-N. Zhang, X. Lu and E.-H. Sheng, RSC Adv., 2015, 5, 29491–29496 RSC; (d) Z. Zhao, H. Yang, Y. Li and X. Guo, Green Chem., 2014, 16, 1274–1281 RSC; (e) X.-J. Yang, B. Chen, L.-Q. Zheng, L.-Z. Wu and C.-H. Tung, Green Chem., 2014, 16, 1082–1086 RSC; (f) X. Liu, S. Ye, H.-Q. Li, Y.-M. Liu, Y. Cao and K.-N. Fan, Catal. Sci. Technol., 2013, 3, 3200–3206 RSC.
- (a) S. Janani, S. Priscilla and V. Anbazhagan, Colloids Surf., B, 2014, 117, 528–533 CrossRef CAS PubMed; (b) A. A. Khan Behlol, S. Sweetha, S. Aravind, V. Ganapathy and V. Anbazhagan, Spectrochim. Acta, Part A, 2014, 130, 54–58 CrossRef PubMed; (c) V. Showmya, V. Ganapathy, E. Elangovan and V. Anbazhagan, RSC Adv., 2014, 4, 15003–15006 RSC; (d) Q. Cui, A. Yashchenok, L. Li, H. Mohwald and M. Bargeheer, Colloids Surf., A, 2015, 470, 108–113 CrossRef CAS.
- T. Aditya, A. Pal and T. Pal, Chem. Commun., 2015, 51, 9410–9431 RSC.
- P. Hervés, M. Pérez-Lorenzo, L. M. Liz-Marzán, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 41, 5577–5587 RSC.
- (a) A. Gangula, R. Podila, M. Ramakrishna, L. Karanam, C. Janardhana and A. M. Rao, Langmuir, 2011, 27, 15268–15274 CrossRef PubMed; (b) L. Ai and J. Jiang, Bioresour. Technol., 2013, 132, 374–377 CrossRef CAS PubMed; (c) A. Zachary, D. Pozun, S. E. Rodenbusch, E. Keller, K. Tran, W. Tang, K. J. Stevenson and G. Henkelman, J. Phys. Chem. C, 2013, 117, 7598–7604 CrossRef PubMed; (d) V. Anbazhagan, K. B. Ayaz Ahmed and S. Janani, Sens. Actuators, B, 2014, 200, 92–100 CrossRef CAS; (e) A. Kumar, K. Smita, E. Sánchez, S. Guerra and L. Cumbal, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2016, 7, 25013 CrossRef; (f) J. Safari, A. E. Najafabadi, Z. Zarnegar and S. F. Masoule, Green Chem. Lett. Rev., 2016, 9, 20–26 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting data, NMR spectra (1H) and UV-vis spectra are provided. See DOI: 10.1039/c6ra21315d |
| This journal is © The Royal Society of Chemistry 2016 |