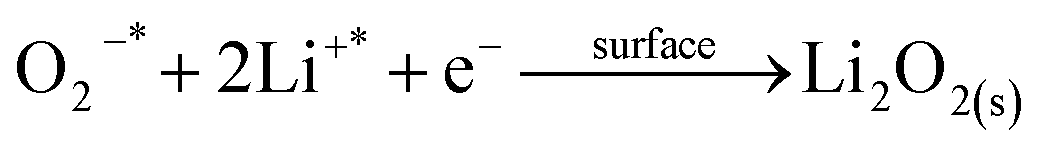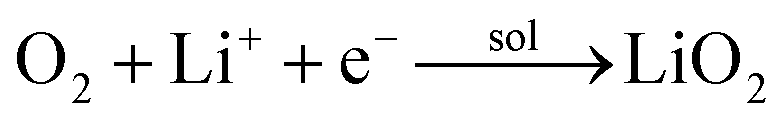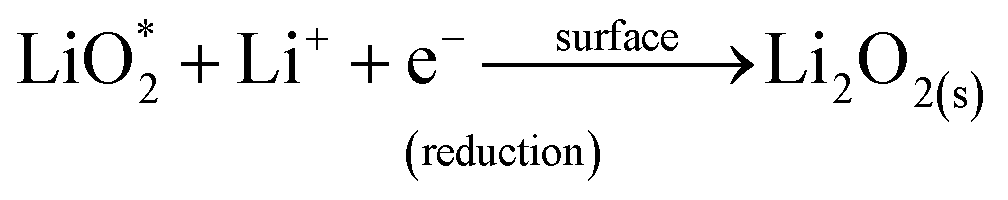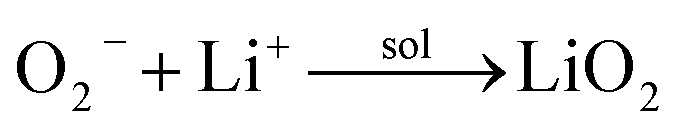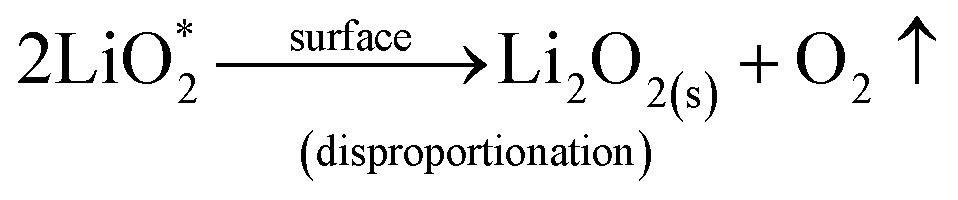DOI:
10.1039/C6RA16874D
(Paper)
RSC Adv., 2016,
6, 103072-103080
Binder-free hierarchically-porous carbon nanofibers decorated with cobalt nanoparticles as efficient cathodes for lithium–oxygen batteries†
Received
30th June 2016
, Accepted 25th September 2016
First published on 11th October 2016
Abstract
The development of efficient cathodes is a great challenge inhibiting the advancement of lithium–oxygen (Li–O2) batteries. In the present study, binder-free, high surface area hierarchically-porous carbon nanofibers decorated with cobalt nanoparticles (Co–PCNF) are investigated as cathodes for Li–O2 batteries. We fabricate the nanofibers using a facile electrospinning technique followed by thermal treatment with in situ incorporation of cobalt nanoparticles. This method provides a free-standing, electron-conducting network with a hierarchical pore structure and effective dispersion of cobalt nanoparticles, which is directly used as a cathode in Li–O2 cells without any binders. Li–O2 cells with Co–PCNF as the cathode exhibit a high initial discharge capacity of 8800 mA h g−1 at the current density of 100 mA g−1, and can be recharged for more than 50 cycles with a limited discharge capacity of 500 mA h g−1. In comparison, porous carbon nanofibers without cobalt provide a discharge capacity of 6670 mA h g−1, and a cycle life of only 35 cycles. The post mortem analysis of discharged cathodes revealed Li2O2 as the major discharge product, and suggested a LiO2-mediated reaction mechanism responsible for the excellent performance of Co–PCNF.
1. Introduction
Rechargeable lithium–air (O2) batteries, with their ultra-high theoretical energy density, about ten-times higher than that of current lithium-ion batteries, are considered to be the most promising of rechargeable battery technologies.1–4 However, the development of these batteries is still at its initial stage, owing to various technical challenges such as high overpotentials, low round-trip efficiency, poor rate capability, and low cycle life. Among various issues affecting Li–O2 battery performance, the low performance of the air cathode has been identified as the dominating factor.5,6 The kinetics of the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) at the cathode govern the overall rate of charge/discharge of the non-aqueous Li–O2 battery and are the limiting factor.7,8 Moreover, the main discharge product of the Li–O2 reaction in a non-aqueous cell, Li2O2,9,10 is insoluble in the electrolyte and insulating in nature. Thus, the cathode should not only provide a three-phase electrochemical interface for the discharge reaction, it should also be able to store the insoluble and non-conductive discharge products as well as catalyze the ORR/OER reactions to reduce the overpotential.11 As a result, the development of efficient cathodes with nano-architectures, large accessible surface area, and bifunctional catalytic activity (for ORR and OER) is a great challenge for high-performance lithium–air batteries.
Carbon based materials have been extensively studied as cathodes for Li–O2 batteries, due to their large surface area, high electronic conductivity, low cost, low density, and easy synthesis of the porous structure.12–19 The formation of passivating layer of Li2CO3 has, however, been observed at the carbon/Li2O2 interfaces, leading to increased overpotentials as well as reduced cycle life. In addition, commonly used polymeric binders (e.g. PVDF, PTFE) have also been shown to be unstable in Li–O2 batteries, blocking active sites and increasing cell impedance.20–23 Recent studies suggest that the formation of Li2CO3 and Li carboxylates in ether electrolytes can arise from the reaction of Li2O2 with carbon during charging at high potentials (>3.5 V) as well as with the electrolyte.24,25 High donor number electrolyte such as DMSO is reported to be more stable than TEGDME.25–27 These studies suggest that the synergy between electrode and electrolyte dictate the performance of Li–O2 cells. Carbon-free cathodes have been developed and reported to improve cycle stability to some extent,28–30 however, at the cost of sacrificing capacity and energy density due to their high mass densities. Thus, modifying carbon materials and developing air cathodes with high stability, optimum porous structure/morphology, binder-free characteristic, and high catalytic activity is critical to the development of Li–O2 batteries.31–33
Carbon nanofibers (CNFs) have been widely used as electrodes in various electrochemical energy storage devices because of their excellent conductivities, large surface areas, and structural stability.34–43 Binder-free electrospun CNFs are potential excellent candidates for air-cathodes in lithium–oxygen batteries for the following reasons: (1) no binders: eliminates the side-reactions due to binders, (2) open macroporous structure: allows for diffusion of electrolyte as well as oxygen, providing sufficient active surface area for the cathode reactions as well as necessary pore volume for storage of discharge products, (3) carbon acts as support as well as a catalyst towards the ORR, and (4) versatility: active metallic nanoparticles can be easily dispersed throughout the nanofiber matrix.
In the present study, binder-free high surface area hierarchically-porous carbon nanofibers modified with cobalt nanoparticles (Co–PCNF) have been studied as efficient cathodes for Li–O2 batteries. The parameters characterizing the performance of the Li–O2 battery using the fabricated materials were evaluated and the post mortem analysis was conducted to understand the corresponding discharge and charge reaction mechanisms. Co–PCNF demonstrated much better performance as cathode in Li–O2 batteries, with high discharge capacity, rechargeability, and rate capability, compared to non-cobalt–carbon cathodes. It was found that the discharge reaction mechanism is altered on introducing cobalt nanoparticles in CNF matrix that greatly improves the rechargeability and cyclability of Co–PCNF.
2. Experimental section
2.1. Materials preparation
Hierarchically-porous carbon nanofiber electrode decorated with cobalt nanoparticles (Co–PCNF) was fabricated by electrospinning a polymer solution consisting of 6.7 wt% polyacrylonitrile (PAN, MW = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma-Aldrich), 10 wt% Nafion (obtained by drying Nafion solution (5 wt%, Ion Power Inc.) at 50 °C for 20 h), 2 wt% cobalt carbonate (cobalt(II) carbonate hydrate, Acros Organics) in N,N-dimethylformamide (DMF, ≥99.8%, Sigma-Aldrich). The electrospinning was carried out at room temperature in a chamber maintained at a relative humidity below 20%. An electric potential of 10–12 kV was applied between the needle containing polymer solution and the Al foil grounded collector kept at a distance of ∼15 cm. The electrospun nanofibers were then stabilized at 280 °C in a convection oven for 5 h and carbonized at 1000 °C for 1 h in a tube furnace under a constant flow of nitrogen (N2) gas (Airgas). An intermediate reduction step at 500 °C to completely reduce cobalt oxides to cobalt was employed during carbonization step, during which N2 was replaced with H2 for 1 h at a constant temperature of 500 °C. Non-cobalt porous carbon nanofibers (PCNF) and non-cobalt non-porous carbon nanofibers (NPCNF) were fabricated similarly by electrospinning a solution of PAN (6.8 wt%), and Nafion (10.2 wt%) in DMF and just PAN (10 wt%) in DMF respectively, followed by pyrolysis (with no intermediate reduction step).
000, Sigma-Aldrich), 10 wt% Nafion (obtained by drying Nafion solution (5 wt%, Ion Power Inc.) at 50 °C for 20 h), 2 wt% cobalt carbonate (cobalt(II) carbonate hydrate, Acros Organics) in N,N-dimethylformamide (DMF, ≥99.8%, Sigma-Aldrich). The electrospinning was carried out at room temperature in a chamber maintained at a relative humidity below 20%. An electric potential of 10–12 kV was applied between the needle containing polymer solution and the Al foil grounded collector kept at a distance of ∼15 cm. The electrospun nanofibers were then stabilized at 280 °C in a convection oven for 5 h and carbonized at 1000 °C for 1 h in a tube furnace under a constant flow of nitrogen (N2) gas (Airgas). An intermediate reduction step at 500 °C to completely reduce cobalt oxides to cobalt was employed during carbonization step, during which N2 was replaced with H2 for 1 h at a constant temperature of 500 °C. Non-cobalt porous carbon nanofibers (PCNF) and non-cobalt non-porous carbon nanofibers (NPCNF) were fabricated similarly by electrospinning a solution of PAN (6.8 wt%), and Nafion (10.2 wt%) in DMF and just PAN (10 wt%) in DMF respectively, followed by pyrolysis (with no intermediate reduction step).
2.2. Li–O2 battery assembly and electrochemical measurements
All the fabricated CNF electrodes are freestanding, and hence were directly applied as cathodes without any further processing. The electrodes were punched to 13 mm diameter circular electrodes with the total (carbon, or carbon and cobalt) loading of 1.2 ± 0.2 mg. The electrochemical performance of all Li–O2 batteries was tested in a coin-cell type system, with holes punched into the positive coin cell cover to allow access to oxygen. The coin cell parts (2032 size, MTI Corp.) and the electrodes were dried under vacuum at 50 °C for at least 1 hour before transferring them into the glovebox (M. Braun Inc.). All the batteries were assembled inside the glovebox under an argon atmosphere with H2O level of ∼1 ppm. A typical Li–O2 coin-cell consisted of a Li foil (14 mm diameter, Alfa Aesar) as anode, 16 mm diameter glass fiber (Whatman GF/D microfiber filter paper) as separator, the 13 mm CNF cathode, a nickel mesh as current collector, and 1 M LiTFSI (Acros Organics), 0.1 M LiNO3 (Aldrich) in tetraethylene glycol dimethyl ether (TEGDME, Aldrich) and dimethyl sulfoxide (DMSO, Alfa Aesar) (1:1 volume ratio) as electrolyte. The coin cells were enclosed in homemade glass cells filled with ultra-high purity grade O2 at ∼1 atm pressure. The galvanostatic charge/discharge tests were carried out using a Maccor battery cycler and cyclic voltammetry (CV) measurements were performed using Gamry's Interface 1000 potentiostat. All the capacities are reported with respect to total cathode (CNF) weight. Before starting the electrochemical tests, all cells were kept at rest (OCV) for 3 h under O2 atmosphere. The cut-off potential for discharge was kept at 2 V and charge at 4.5 V for all charge–discharge tests, except for CV which was carried out between 2.2–4.4 V at 0.1 mV s−1 scan rate.
2.3. Characterization
A Zeiss Supra 50VP scanning electron microscope (SEM) fitted with Oxford energy-dispersive X-ray microanalysis (EDAX) detector was used to study the morphology and elemental mapping of electrodes before and after discharge. Transmission electron microscopy (TEM) was performed using a JEOL JEM2100 microscope at an accelerated voltage of 120 kV. The nitrogen adsorption–desorption isotherms were measured at 77 K with an automated gas sorption analyzer (AutoSorb iQ2, Quantachrome Instruments). The surface area and pore volume were assessed by a 7-point model Brunauer–Emmett–Teller (BET) method. The pore-size distributions were calculated using the Barrett–Joyner–Halenda (BJH) method for a broad pore size distribution and density functional theory (DFT) model for micro/mesopore analysis. For XRD analysis, the samples were characterized using a Rigaku SmartLab X-ray diffractometer in Bragg–Brentano mode (Cu Kα radiation, voltage 40 kV, current 40 mA, scanning range 20–80° and step size of 0.02°). X-ray photoelectron spectroscopy (XPS) measurements were performed on Physical Electronics VersaProbe 5000 spectrometer using monochromated Al Kα excitation source. For post mortem analysis, the Li–O2 cells were opened inside the glovebox after electrochemical tests to recover cathode for post mortem analysis. The samples for SEM were prepared inside the glovebox and transported through an air-tight box to the SEM chamber. The XRD samples were also prepared in the glovebox and sealed with Kapton tape for analysis.
3. Results and discussion
3.1. Synthesis and characterization of Co–PCNF
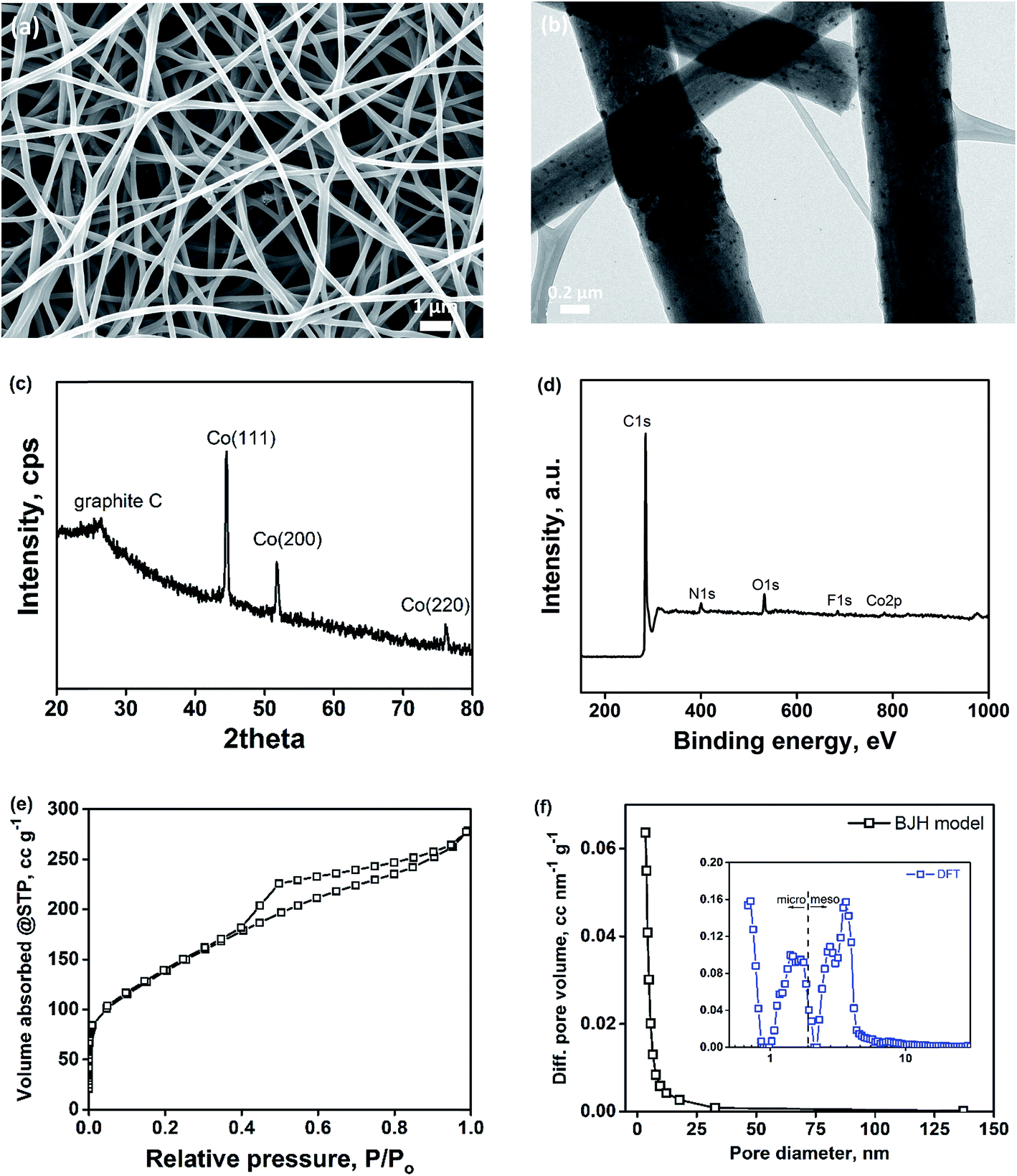
Binder-free porous carbon nanofibers decorated with cobalt nanoparticles (Co–PCNF) were fabricated by electrospinning a polymer solution of polyacrylonitrile (PAN, carbon precursor), Nafion (sacrificial polymer to develop intra-fiber pores), and cobalt carbonate (cobalt precursor for in situ synthesis of cobalt) in DMF. The resulting non-woven mat of electrospun nanofibers was pyrolyzed to form Co–PCNF (Fig. 1a) as described in the Experimental section. The as-electrospun nanofiber mats are free-standing both before and after pyrolysis as shown in Fig. S1.† During pyrolysis, Nafion decomposes out of the carbon matrix forming pores,34,44 while still retaining the free-standing nanofibrous assembly and cobalt carbonate was reduced to cobalt nanoparticles that are uniformly distributed throughout the nanofibers as shown in the TEM image in Fig. 1b. The uniform distribution of cobalt nanoparticles within the nanofibers is further verified from the EDX mapping of cobalt in Co–PCNF (Fig. S2†). XRD spectrum (Fig. 1c) shows the major peaks corresponding to the cubic cobalt phase (PDF: 01-077-7456). The XPS analysis of the Co–PCNF nanofibers shows the presence of C, N, O, F, and Co elements on its surface (Fig. 1d). While nitrogen peak arises from the N-rich PAN, oxygen peak arises from the stabilization step (oxidation of PAN nanofibers in air) during pyrolysis, and fluorine peak is the residual F from Nafion. As indicated by the previous reports,45–49 electronegative heteroatom doping on carbon can modify the chemical and electronic structure of the carbon, thereby enhancing its electrocatalytic activity, as discussed later. Fig. 1e shows the nitrogen adsorption isotherms and Fig. 1f shows the pore size distribution (PSD) for Co–PCNF. The surface area, micropore volume, and total pore volume of Co–PCNF are 490.3 m2 g−1, 0.184 cm3 g−1, and 0.4313 cm3 g−1, respectively. The broad PSD characterized by the BJH model shows the highly meso/macroporous structure of the Co–PCNF. The DFT model (inset of Fig. 1f) reveals the micropore mode of 0.7 nm, and the mesopore mode of 3.63 nm. Thus, the porosity analysis demonstrates the hierarchical micro/meso/macroporous structure of Co–PCNF.
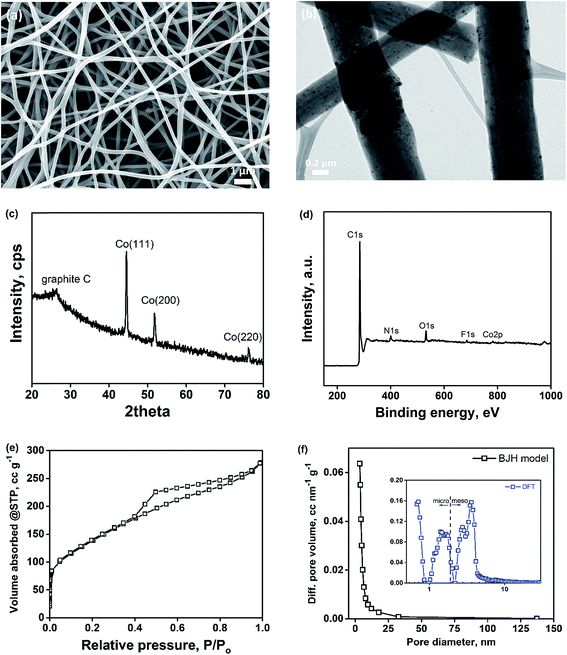 |
| | Fig. 1 Characterization of Co–PCNF. (a) SEM image; (b) TEM image; (c) XRD spectrum; (d) XPS elemental spectrum; (e) nitrogen-isotherm; (f) pore-size distribution. | |
3.2. Electrochemical performance in Li–O2 battery and post mortem analysis of the cathode
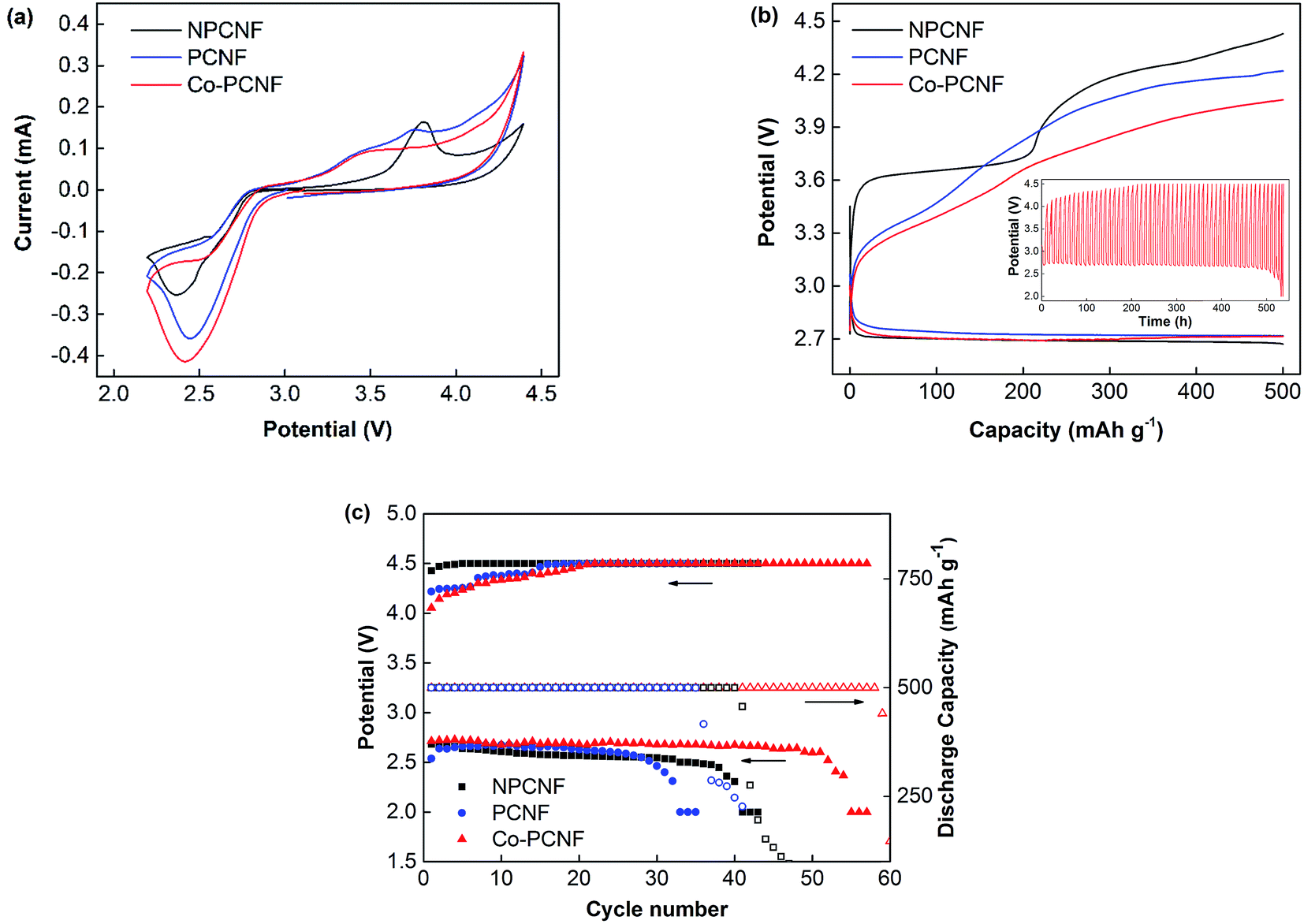
For a better understanding of the performance of Co–PCNF as a cathode in Li–O2 battery, its performance is compared with that of porous (PCNF) and non-porous carbon nanofibers (NPCNF), without any cobalt. PCNF, like Co–PCNF exhibits a high surface area of 563.20 m2 g−1, while NPCNF has a low surface area of only 29.15 m2 g−1. Fig. 2a shows the CV curves at 0.1 mV s−1 to investigate the electrocatalytic activity of NPCNF, PCNF, and Co–PCNF cathodes in aprotic Li–O2 batteries. PCNF and Co–PCNF show remarkably improved onset discharge and charge potentials and higher current density than NPCNF, indicating their higher electrocatalytic activity. While NPCNF shows a distinguishable anodic peak corresponding to oxidation of Li2O2, the anodic curves for PCNF and Co–PCNF show gradual oxidation starting at a lower potential.
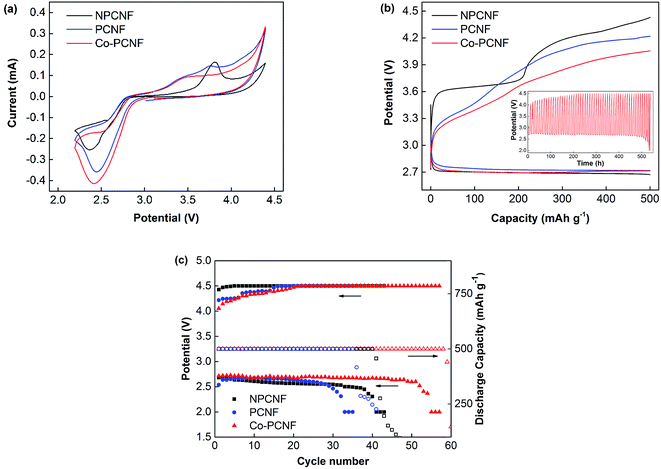 |
| | Fig. 2 Electrochemical performance of Li–O2 batteries with NPCNF, PCNF and Co–PCNF cathodes. (a) CV curves at 0.1 mV s−1 between 2.2 and 4.4 V; (b) discharge–charge curves at 100 mA g−1 with limited capacity of 500 mA h g−1, inset: galvanostatic cycling curves of Co–PCNF at 100 mA g−1; (c) cycling performance and terminal charge–discharge potentials. | |
Fig. 2b compares the initial discharge–charge profiles of NPCNF, PCNF, and Co–PCNF electrode cells with a limited capacity of 500 mA h g−1 at 100 mA g−1 current density. The onset discharge potential follows a similar trend as seen in the CV curves with PCNF and Co–PCNF showing a slightly lower onset potential than NPCNF. Moreover, all samples show a similar discharge potential plateau at ∼2.72 V. During charging, the difference in the observed potential between the three samples is much more pronounced. Co–PCNF showed the lowest charging potential beginning at ∼3.0 V and attaining a charge of 500 mA h g−1 at 4.05 V. PCNF also showed a similar onset charging potential of ∼3.0 V, with the end potential being 4.2 V. NPCNF, however, showed the highest onset charging potential of ∼3.3 V along with a prominent voltage plateau at an average potential of 3.65 V, and end potential of 4.43 V. Thus, Co–PCNF exhibits the lowest overpotential among the three cathodes, lower by 150 mV from PCNF and 380 mV from NPCNF. The inset curve compares the charge–discharge profiles for the three cathodes during cycling. The cycling stability of the cathodes is further shown in Fig. 2c where terminal charge/discharge potential and discharge capacity are shown as a function of cycle number. NPCNF cathode is able to achieve 41 cycles and PCNF 35 cycles before capacity fading starts. Co–PCNF showed the best performance with no capacity fading for over 50 (58) cycles and with almost constant discharge potential of 2.68 V, indicating better reversibility of Co–PCNF electrode. Even though PCNF showed lower (better) overpotential (Fig. 2b), the cycle life of PCNF is lower than that of NPCNF, while Co–PCNF shows the best cycle life. This indicates that porous carbon defects (present in PCNF) are unstable on cycling, however, the stability improves on modifying the carbon surface with cobalt.
We then conducted the post mortem analysis of the discharged cathodes. Fig. 3 shows the SEM images for each of the three cathodes in the pristine state, after first discharge and after the first recharge. For all three carbon nanofiber electrodes, discharge products are formed conformally to the nanofibrous structure, and they disappear on charging, suggesting carbon nanofibers serve as excellent sites for discharge product accumulation, and discharge and charge reactions. While NPCNF and PCNF show a toroidal product morphology, Co–PCNF exhibits two different morphologies of discharge products—amorphous nanosheets dispersed loosely around electrode surface, and toroids. The toroidal product formation is consistent with the earlier reports on carbon-based electrodes,13,20,50 which suggest that the product, Li2O2 nucleates and grows on the surface and in the solution simultaneously as the superoxide can both dissolve in the electrolyte as well as get trapped on the carbon defect sites (see eqn (1) and (2)).
| |
 | (1) |
| |
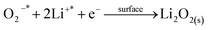 | (2) |
where, the subscript “sol” stands for the solution phase, “*” sign indicates adsorbed species, “s” stands for solid film. The presence of an additional morphology on Co–PCNF along with the toroids indicates an additional growth pathway of Li
2O
2 at Co–PCNF influenced by the presence of cobalt nanoparticles. As indicated by previous reports,
33,51,52 Co nanoparticles can possibly lower the adsorption energy of LiO
2 inducing surface nucleation process. Thus, we expect that the formation of the nanosheet-type Li
2O
2 on Co–PCNF occurs
via the disproportionation/reduction of LiO
2 through the surface nucleation process, favored by the adsorption of LiO
2 by Co nanoparticles. The possible reaction mechanisms are given below:
53| |
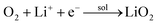 | (3) |
| |
 | (4) |
or,
| |
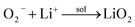 | (5) |
| |
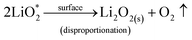 | (6) |
 |
| | Fig. 3 SEM images of (a)–(c) NPCNF, (d)–(f) PCNF, and (g)–(i) Co–PCNF cathodes at various stages: pristine (a, d and g), after first discharge (b, e and h) and after recharge (c, f and i). | |
The average thickness of Li2O2 toroid particles formed around the nanofibers was calculated by subtracting the average diameter of nanofibers from the average toroid diameter and it decreases in the order: NPCNF (0.245 μm) > PCNF (0.230 μm) > Co–PCNF (0.106 μm). The remarkably small Li2O2 toroid particles formed for Co–PCNF can potentially enhance the reactivity and electrode kinetics,54 leading to lower polarization and better rechargeability for Co–PCNF based Li–O2 cells as seen in Fig. 2.
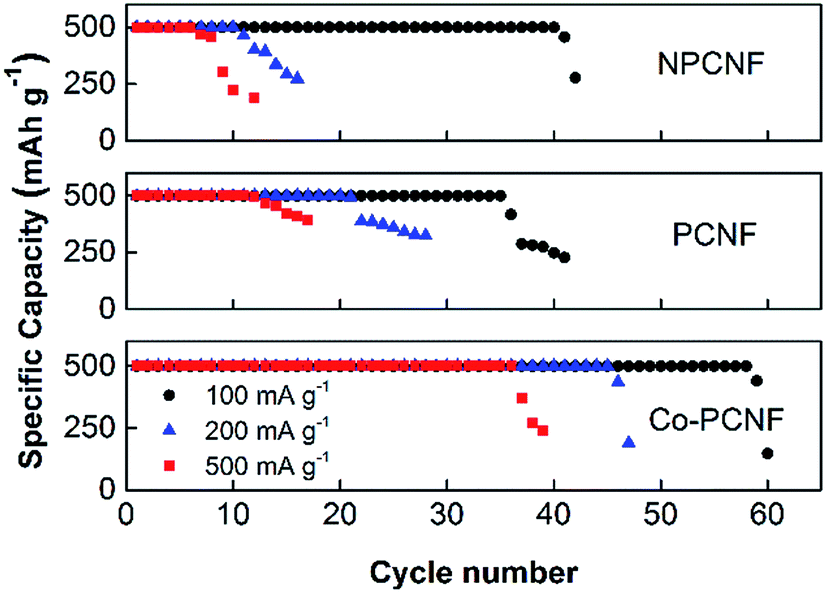
The rate performance of the electrodes was then investigated at varying current densities of 100, 200 and 500 mA g−1 and the corresponding cycling response is shown in Fig. 4. The initial charge–discharge curves for the three electrodes at different current densities are shown in Fig. S3,† and the performance follows the order: Co–PCNF > PCNF > NPCNF. At a high current density of 500 mA g−1, Co–PCNF can still achieve 36 cycles compared to only 11 cycles by PCNF. Moreover, the increase in overpotential with increasing current densities is also the lowest for Co–PCNF (Fig. S3†), indicating faster reaction kinetics at Co–PCNF.
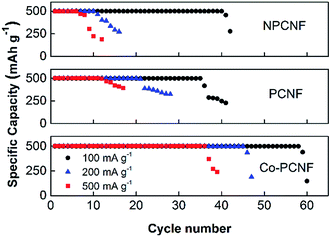 |
| | Fig. 4 Rate performance of NPCNF, PCNF and Co–PCNF at various current densities. | |
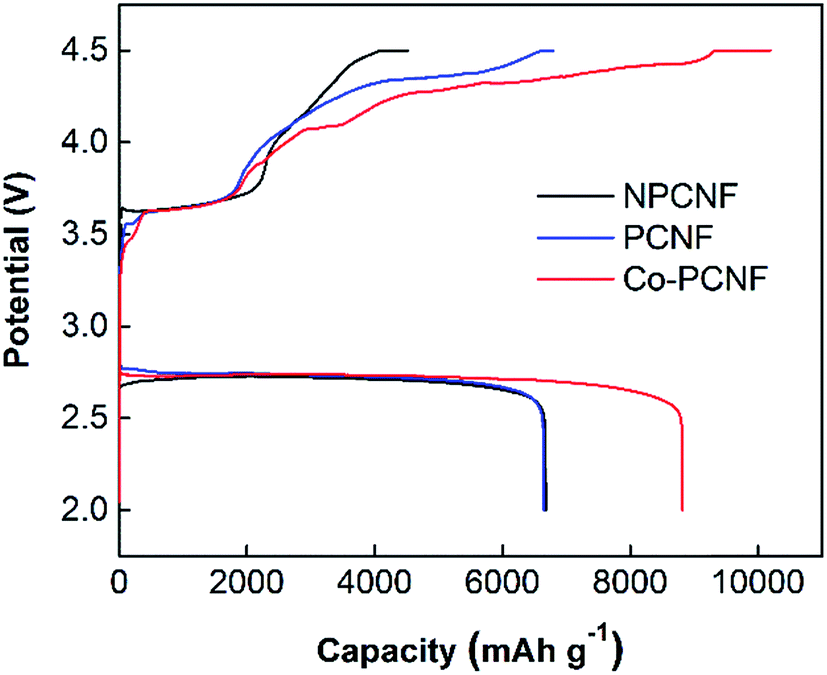
To further explore the electrochemical performance of the three electrodes, deep discharge and charge at 100 mA g−1 with cut-off discharge and charge potentials of 2.0 and 4.5 V vs. Li/Li+ respectively, was investigated as shown in Fig. 5. The deep discharging provides the maximum achievable capacity of the cathode, however, it also leads to higher cathode polarization due to thicker discharge product deposits. The formation of thicker product deposits that are insulating in nature is expected to increase the charging potentials, as also observed here (comparing Fig. 5 and 2b). Co–PCNF exhibits the largest discharge capacity of 8800 mA h g−1, followed by PCNF (6670 mA h g−1) and NPCNF (6650 mA h g−1). The similar discharge capacity achieved for NPCNF and PCNF indicates that higher surface area generated due to the presence of small pores (micro and mesopores), does not necessarily augment the discharge capacity. A recent study55 showed through XPS depth-profiling that small pores could impede the electrolyte (and hence dissolved O2 and Li+) diffusion leading to clogging of pores rather than complete filling, thereby reducing the specific capacity of the electrode, while macropores allow for a constant oxygen and Li+ feed inside the electrode enabling discharge and charge reactions. Moreover, the larger discharge capacity of Co–PCNF indicates the more efficient deposition of discharge products, also demonstrated by the smaller toroids and amorphous nanosheets.
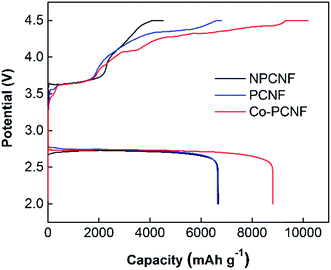 |
| | Fig. 5 Comparison of deep discharge–charge curves for NPCNF, PCNF and Co–PCNF electrodes at 100 mA g−1 with the cut-off potentials of 2.0 and 4.5 V. | |
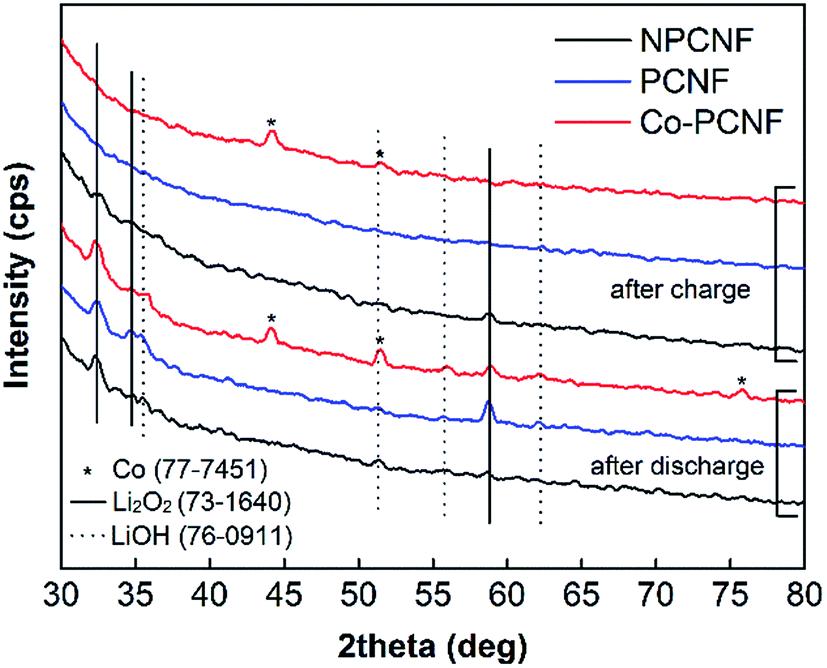
The discharge products after deep discharge and charge were further characterized using post mortem XRD and SEM-EDX. As shown in Fig. 6, Li2O2 is the main discharge product, with its diffraction peaks clearly present on all three electrodes, along with small LiOH peaks. LiOH is a side-product possibly formed through the side-reaction with the traces of moisture possibly present in O2 feed gas. On charging, these peaks disappear completely for PCNF and Co–PCNF, with small Li2O2 peaks still remaining for NPCNF, indicating inefficient rechargeability for the latter. This is also corroborated by the lower charge capacity achieved for NPCNF (Fig. 5) and O-distribution from SEM-EDX images shown in Fig. 7a–f. The higher intensity of O (red dots) that characterizes the discharge product is seen in the discharged cathodes and the intensity greatly diminishes on charging. It is to be noted that some oxygen functionalities are present in all pristine carbon cathodes owing to the polyacrylonitrile pyrolysis process therefore oxygen signal in SEM-EDX doesn't completely disappear after re-charge.
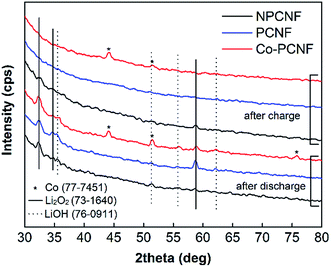 |
| | Fig. 6 XRD patterns of discharged and re-charged NPCNF, PCNF, and Co–PCNF electrodes. | |
 |
| | Fig. 7 Element map of O (red) in (a) and (b) NPCNF, (c) and (d) PCNF, and (e) and (f) Co–PCNF cathodes at various stages: after deep discharge (a, c and e) and after deep charge (b, d and f) using SEM-EDX. | |
The excellent performance of Co–PCNF even at higher current densities is attributed to the synergistic effect of cobalt nanoparticles and N- and F-doped carbon that serve as active catalytic sites as well as provide faster electronic interaction. The role of these active sites is discussed below.
3.3. Elucidation of mechanism
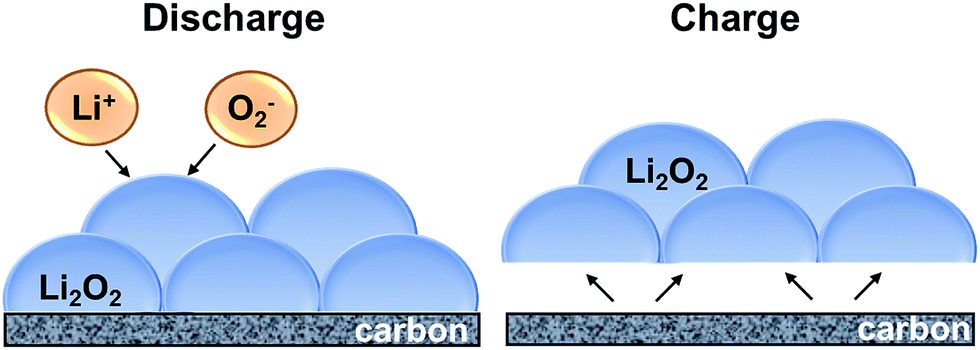
Here, we propose the reaction mechanism for the discharge and charge reactions and the potential role of N-and F-doped carbon defect sites and cobalt nanoparticles based on the above results and literature reports. A schematic of the proposed discharge and charge mechanism is shown in Fig. 8. Discharge reaction is driven by the reaction of dissolved Li+ and O2− ions in the electrolyte solution at the electrode/Li2O2–electrolyte interface, which in turn is governed by the diffusion of Li+ and O2− through the electrolyte and the transport of electrons through the deposited Li2O2 product.56 Although bulk Li2O2 has poor electron conductivity, electron transfer can occur through the surface defects/half-metallic states of Li2O2 that allow for thicker Li2O2 deposits and thereby, higher discharge capacity.57,58 Given that the reaction continues to take place at the product–electrolyte interface, the discharge takes place at a constant potential (Fig. 2b), and competitive discharge capacity is achieved (Fig. 5) for all three macroporous carbon nanofiber electrodes.
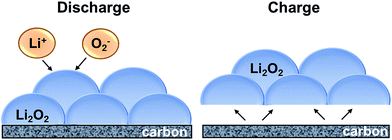 |
| | Fig. 8 Schematic of discharge and charge mechanism for Li2O2 formation and dissociation. | |
The oxygen evolution (charging) reaction, on the other hand, takes place at the electrode–Li2O2 interface, instead of Li2O2–electrolyte interface.56 A good binding force between the reaction product and the electrode is thus necessary for efficient charging, and hence is influenced by the surface chemistry and electrocatalytic activity of the electrode. Various studies on heteroatom-doped carbon-based materials have shown to exhibit improved electrochemical performance in lithium–oxygen batteries. Yan et al.59 using DFT calculations, showed that electronegative atom-doping (such as N) could not only enhance the adsorption of O2 molecule due to energetically stable charge transfer, but also decrease the energy barrier of O2 dissociation from 2.39 eV to 1.20 eV. The N- and F-doped carbon defect sites in PCNF and Co–PCNF are therefore expected to enhance adsorption of O2/O2− and thereby lower the overpotentials, as observed in Fig. 2b.
Note that while the strong adsorption of oxygen due to surface functionalities is expected to lower the onset discharge potential, it is also known that the strongly adsorbed superoxide (O2−) could react with carbon defects as well as the electrolyte to form unwanted Li2CO3.60 The build-up of Li2CO3 would polarize the electrode surface, increasing the charging potentials on cycling, and reduce the cycle life as seen in Fig. 2c. PCNF with higher carbon defects (owing to intra-fiber porosity) shows poor reversibility compared to NPCNF. However, interestingly, Co–PCNF with similar carbon defects as PCNF shows better cyclability. This potentially arises from the presence of cobalt nanoparticles. As discussed earlier, cobalt nanoparticles could lower the activation energy of adsorption of LiO2, and hence possibly induce an alternative reaction mechanism for the formation of product via LiO2 reduction/disproportionation at the metal surfaces instead of carbon surfaces. Further, Nazar et al.51 have proposed that metal/metal oxides (including cobalt-based) promote the mass transport of LixO2 species, since metal/metal oxide surfaces are less “sticky” than carbon surfaces riddled with dangling bonds that strongly bind to superoxide. Thus, we deduce that the presence of cobalt nanoparticles in Co–PCNF not only lowers the charging overpotentials, but also improves the cycle life via LiO2-mediated reaction mechanism.
4. Conclusions
Cobalt embedded hierarchically-porous carbon nanofibers are presented as efficient binder-free cathodes for lithium–oxygen batteries. They demonstrate much better discharge capacity, cycling performance and rate capability than non-cobalt carbon nanofibers. Their performance is attributed to the binder-free, inter-connected hierarchical pore structure of carbon nanofibers with effective dispersion of cobalt nanoparticles, providing fast electron transport as well as fast ion-diffusion. Moreover, the post mortem analysis showed that the presence of cobalt nanoparticles altered the reaction mechanism, stabilizing carbon surface via LiO2-mediated reactions, enhancing the reversibility and cycle life.
Acknowledgements
This work is supported by the National Science Foundation under grant number CBET-1236466. The authors would like to thank Dr Sheng-Heng Chung and Prof. Arumugam Manthiram for porosimetry measurements, Dr Craig Johnson for TEM and Dr Dmitri Barbash for XPS. The authors are grateful to Centralized Research Facility of Drexel University for instrumentation support.
References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 172 CrossRef CAS.
- N. Imanishi and O. Yamamoto, Mater. Today, 2014, 17, 24–30 CrossRef CAS.
- L. Mai, X. Tian, X. Xu, L. Chang and L. Xu, Chem. Rev., 2014, 114, 11828–11862 CrossRef CAS PubMed.
- Z. Ma, X. Yuan, L. Li, Z.-F. Ma, D. P. Wilkinson, L. Zhang and J. Zhang, Energy Environ. Sci., 2015, 8, 2144–2198 CAS.
- A. Débart, J. Bao, G. Armstrong and P. G. Bruce, J. Power Sources, 2007, 174, 1177–1182 CrossRef.
- F. Cheng and J. Chen, Nat. Chem., 2012, 4, 962–963 CrossRef CAS PubMed.
- K. M. Abraham and Z. Jiang, J. Electrochem. Soc., 1996, 143, 1–5 CrossRef CAS.
- T. Ogasawara, A. Débart, M. Holzapfel, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 1390–1393 CrossRef CAS PubMed.
- J. Wang, Y. Li and X. Sun, Nano Energy, 2013, 2, 443–467 CrossRef CAS.
- X.-h. Yang, P. He and Y.-y. Xia, Electrochem. Commun., 2009, 11, 1127–1130 CrossRef CAS.
- R. R. Mitchell, B. M. Gallant, C. V. Thompson and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 2952–2958 CAS.
- Y. Li, J. Wang, X. Li, J. Liu, D. Geng, J. Yang, R. Li and X. Sun, Electrochem. Commun., 2011, 13, 668–672 CrossRef CAS.
- J. Xiao, D. Mei, X. Li, W. Xu, D. Wang, G. L. Graff, W. D. Bennett, Z. Nie, L. V. Saraf, I. A. Aksay, J. Liu and J.-G. Zhang, Nano Lett., 2011, 11, 5071–5078 CrossRef CAS PubMed.
- H.-G. Jung, J. Hassoun, J.-B. Park, Y.-K. Sun and B. Scrosati, Nat. Chem., 2012, 4, 579–585 CrossRef CAS PubMed.
- Z. Guo, D. Zhou, X. Dong, Z. Qiu, Y. Wang and Y. Xia, Adv. Mater., 2013, 25, 5668–5672 CrossRef CAS PubMed.
- J. Li, Y. Zhang, W. Zhou, H. Nie and H. Zhang, J. Power Sources, 2014, 262, 29–35 CrossRef CAS.
- J. Xie, X. Yao, Q. Cheng, I. P. Madden, P. Dornath, C. C. Chang, W. Fan and D. Wang, Angew. Chem., 2015, 127, 4373–4377 CrossRef.
- R. Black, S. H. Oh, J.-H. Lee, T. Yim, B. Adams and L. F. Nazar, J. Am. Chem. Soc., 2012, 134, 2902–2905 CrossRef CAS PubMed.
- W. Xu, J. Hu, M. H. Engelhard, S. A. Towne, J. S. Hardy, J. Xiao, J. Feng, M. Y. Hu, J. Zhang and F. Ding, J. Power Sources, 2012, 215, 240–247 CrossRef CAS.
- R. Younesi, M. Hahlin, M. Treskow, J. Scheers, P. Johansson and K. Edström, J. Phys. Chem. C, 2012, 116, 18597–18604 CAS.
- R. Younesi, S. Urbonaite, K. Edström and M. Hahlin, J. Phys. Chem. C, 2012, 116, 20673–20680 CAS.
- B. D. McCloskey, A. Speidel, R. Scheffler, D. C. Miller, V. Viswanathan, J. S. Hummelshøj, J. K. Nørskov and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 997–1001 CrossRef CAS PubMed.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, J. Am. Chem. Soc., 2012, 135, 494–500 CrossRef PubMed.
- C. O. Laoire, S. Mukerjee, K. M. Abraham, E. J. Plichta and M. A. Hendrickson, J. Phys. Chem. C, 2010, 114, 9178–9186 CAS.
- F. S. Gittleson, R. C. Sekol, G. Doubek, M. Linardi and A. D. Taylor, Phys. Chem. Chem. Phys., 2014, 16, 3230–3237 RSC.
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS PubMed.
- G. Zhao, J. Lv, Z. Xu, L. Zhang and K. Sun, J. Power Sources, 2014, 248, 1270–1274 CrossRef CAS.
- C. Shen, Z. Wen, F. Wang, X. Huang, K. Rui and X. Wu, RSC Adv., 2016, 6, 16263–16267 RSC.
- J.-L. Shui, N. K. Karan, M. Balasubramanian, S.-Y. Li and D.-J. Liu, J. Am. Chem. Soc., 2012, 134, 16654–16661 CrossRef CAS PubMed.
- J. J. Xu, Z. L. Wang, D. Xu, L. L. Zhang and X. B. Zhang, Nat. Commun., 2013, 4, 2438 Search PubMed.
- P. Zhang, R. Wang, M. He, J. Lang, S. Xu and X. Yan, Adv. Funct. Mater., 2016, 26, 1354–1364 CrossRef CAS.
- C. Tran and V. Kalra, J. Power Sources, 2013, 235, 289–296 CrossRef CAS.
- C. Tran, R. Singhal, D. Lawrence and V. Kalra, J. Power Sources, 2015, 293, 373–379 CrossRef CAS.
- B. Zhang, F. Kang, J.-M. Tarascon and J.-K. Kim, Prog. Mater. Sci., 2016, 76, 319–380 CrossRef CAS.
- L.-F. Chen, X.-D. Zhang, H.-W. Liang, M. Kong, Q.-F. Guan, P. Chen, Z.-Y. Wu and S.-H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS PubMed.
- B.-W. Huang, X.-Z. Liao, H. Wang, C.-N. Wang, Y.-S. He and Z.-F. Ma, J. Electrochem. Soc., 2013, 160, A1112–A1117 CrossRef CAS.
- Y. J. Kim, H. Lee, D. J. Lee, J. K. Park and H. T. Kim, ChemSusChem, 2015, 8, 2496–2502 CrossRef CAS PubMed.
- B. N. Joshi, S. An, H. S. Jo, K. Y. Song, H. G. Park, S. Hwang, S. S. Al-Deyab, W. Y. Yoon and S. S. Yoon, ACS Appl. Mater. Interfaces, 2016, 8, 9446–9453 CAS.
- L. Xia, S. Wang, G. Liu, L. Ding, D. Li, H. Wang and S. Qiao, Small, 2015, 12, 853–859 CrossRef PubMed.
- S. H. Chung, R. Singhal, V. Kalra and A. Manthiram, J. Phys. Chem. Lett., 2015, 6(12), 2163–2169 CrossRef CAS PubMed.
- R. Singhal and V. Kalra, J. Mater. Chem. A, 2015, 3(1), 377–385 CAS.
- C. Tran and V. Kalra, Soft Matter, 2013, 9, 846–852 RSC.
- X. Sun, P. Song, Y. Zhang, C. Liu, W. Xu and W. Xing, Sci. Rep., 2013, 3, 2505 Search PubMed.
- X. J. Sun, Y. W. Zhang, P. Song, J. Pan, L. Zhuang, W. L. Xu and W. Xing, ACS Catal., 2013, 3, 1726–1729 CrossRef CAS.
- D. S. Geng, Y. Chen, Y. G. Chen, Y. L. Li, R. Y. Li, X. L. Sun, S. Y. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760–764 CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- D. Shin, B. Jeong, B. S. Mun, H. Jeon, H.-J. Shin, J. Baik and J. Lee, J. Phys. Chem. C, 2013, 117, 11619–11624 CAS.
- K.-H. Xue, E. McTurk, L. Johnson, P. G. Bruce and A. A. Franco, J. Electrochem. Soc., 2015, 162, A614–A621 CrossRef CAS.
- R. Black, J. H. Lee, B. Adams, C. A. Mims and L. F. Nazar, Angew. Chem., 2013, 125, 410–414 CrossRef.
- B. G. Kim, H. J. Kim, S. Back, K. W. Nam, Y. Jung, Y. K. Han and J. W. Choi, Sci. Rep., 2014, 4, 4225 Search PubMed.
- D. Zhai, H.-H. Wang, J. Yang, K. C. Lau, K. Li, K. Amine and L. A. Curtiss, J. Am. Chem. Soc., 2013, 135, 15364–15372 CrossRef CAS PubMed.
- Y. Hu, X. Han, F. Cheng, Q. Zhao, Z. Hu and J. Chen, Nanoscale, 2013, 6, 177–180 RSC.
- I. Bardenhagen, M. Fenske, D. Fenske, A. Wittstock and M. Bäumer, J. Power Sources, 2015, 299, 162–169 CrossRef CAS.
- A. Kushima, T. Koido, Y. Fujiwara, N. Kuriyama, N. Kusumi and J. Li, Nano Lett., 2015, 15, 8260–8265 CrossRef CAS PubMed.
- M. D. Radin, J. F. Rodriguez, F. Tian and D. J. Siegel, J. Am. Chem. Soc., 2011, 134, 1093–1103 CrossRef PubMed.
- M. D. Radin and D. J. Siegel, Energy Environ. Sci., 2013, 6, 2370–2379 CAS.
- H. Yan, B. Xu, S. Shi and C. Ouyang, J. Appl. Phys., 2012, 112, 104316 CrossRef.
- D. M. Itkis, D. A. Semenenko, E. Y. Kataev, A. I. Belova, V. S. Neudachina, A. P. Sirotina, M. Hävecker, D. Teschner, A. Knop-Gericke and P. Dudin, Nano Lett., 2013, 13, 4697–4701 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra16874d |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.