
Design of boronic acid-attributed carbon dots on inhibits HIV-1 entry†
Abstract
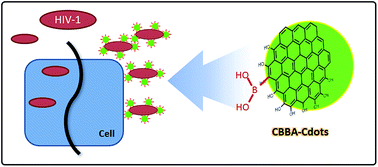
The development of gp120 targeted human immunodeficiency virus (HIV) drug has improved antiretroviral therapies owing to its effects on attachment to target cells. Some currently available antiretroviral therapies that act by inhibiting viral infection still are limited on the toxicity issues; therefore, novel approaches to treat and prevent HIV infections are still needed. Herein, we introduce carbon dot nanoparticles as a new strategy for preventing HIV-1 infection via interaction with gp120 and subsequent elimination of target cell interaction. Carbon dots (diameter: ∼2 nm) exhibiting a graphene-like structure were prepared by pyrolysis of citric acid and further associated with boronic acid-containing molecules. Specific peaks from Fourier transform infrared spectroscopy and X-ray photoelectron spectroscopy analysis indicated successful modification of the carbon dots by boronic acid. The lower cytotoxic effects of this carbon-based material were evaluated using WST-1 assays. The existence of boronic acid moieties on the edge of carbon dots enhanced the inhibitory activity by suppressing syncytium formation. These findings provide a basis for further studies of carbon dot-based applications in HIV prevention and therapy.
 Please wait while we load your content...
Please wait while we load your content...

